Two-in-one: concomitant diffuse large B-cell lymphoma and cavernous haemangioma within the same orbit
Hong Kong Med J 2024 Jun;30(3):249 | Epub 4 Jun 2024
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Two-in-one: concomitant diffuse large B-cell lymphoma and cavernous haemangioma within the same orbit
Kenneth KH Lai, MB, ChB, AFCOphth1; Tiffany Ong, MB, ChB1; Tiffany HT Chan, MB, ChB2; Edwin Chan, FCOphth1; Andrew KT Kuk, FCOphth1
1 Department of Ophthalmology, Tung Wah Eastern Hospital, Hong Kong SAR, China
2 Department of Anatomical and Cellular Pathology, Pamela Youde Nethersole Eastern Hospital, Hong Kong SAR, China
Corresponding author: Dr Andrew KT Kuk (aktkuk@hku.hk)
 Full paper in PDF
Full paper in PDF
Orbital tumours encompass a wide range of benign
and malignant space occupying lesions that may
arise primarily from the orbit or have spread
from other sites in the body. They are rare with an
incidence of 1 in every 100 000 and may lead to
devastating complications of which mechanical
compression causing optic neuropathy is the most
important.1 Multiple orbital tumours of the same
orbit are even rarer with most reported cases being
benign homologous tumours such as cavernous
haemangiomas or myxofibrosarcomas.2 3 A 58-year-old
ethnic Han Chinese male presented in December
2021 with a 1-month history of right eye proptosis.
He had no clinical sign of optic neuropathy.
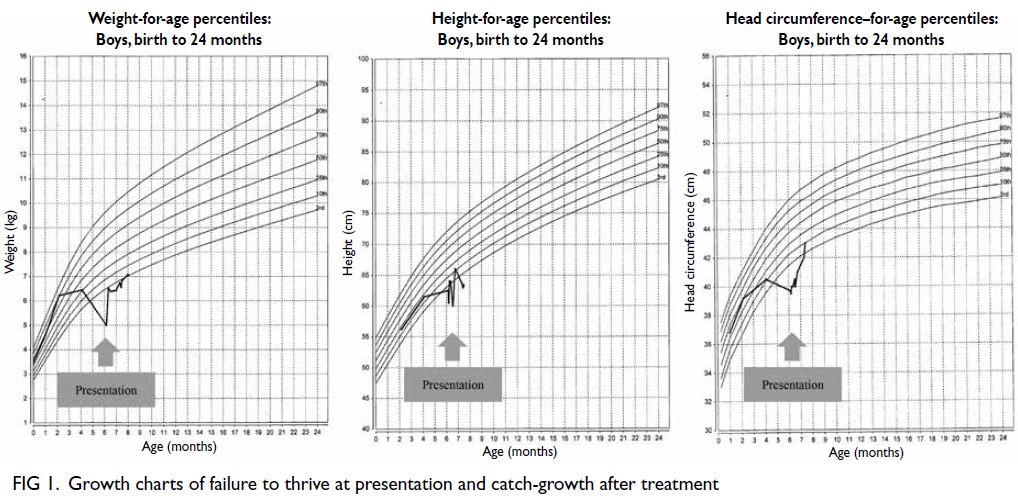
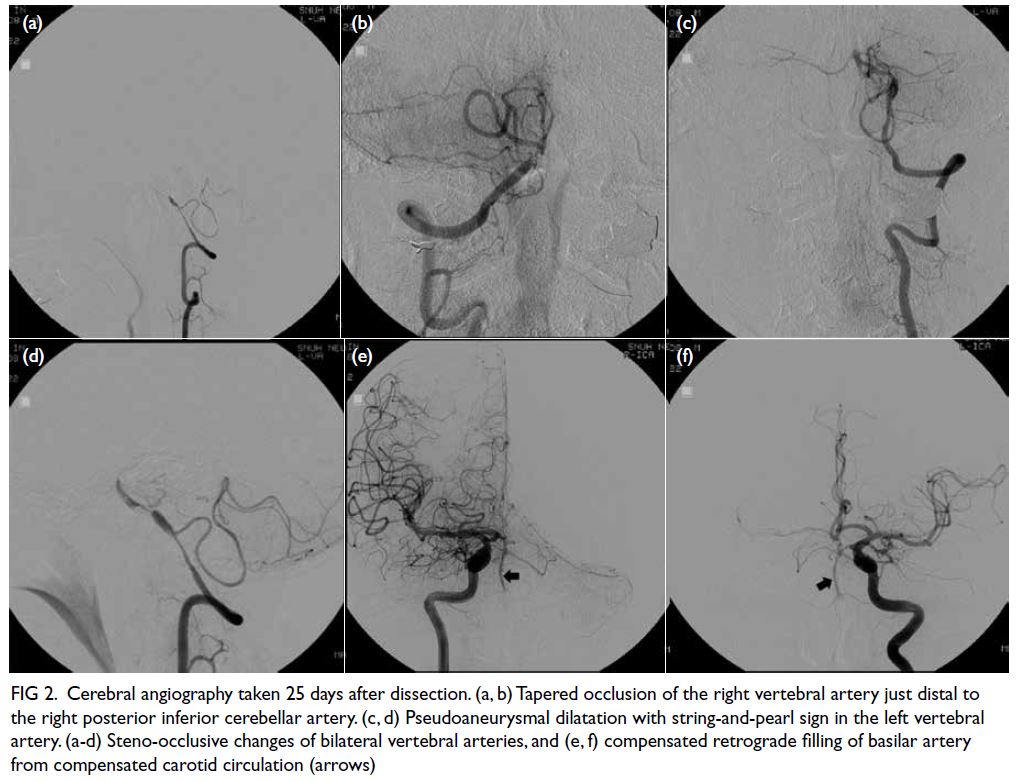
Computed tomography of the orbit revealed right
axial proptosis and two separate lesions in the
right orbit. One lesion appeared infiltrative and
measured 2.2 × 1.5 × 1.7 cm3 (anteroposterior × transverse × longitudinal) at the extraconal space
with retrobulbar extension between the lamina
papyracea and the medial rectus. The other was an
encapsulated mass with regular border located in
the superolateral intraconal region measuring 1.9 × 1.7 × 2.0 cm3 and abutting the optic nerve. Both
lesions enhanced mildly with intravenous contrast (Fig 1). Blood results revealed normal thyroid
function and immunoglobulin G4 and white blood
cell levels. Based on the distinguishing radiological
features of each lesion, we performed a two-stage
surgery for theranostic reasons. An incisional biopsy
of the medial infiltrative lesion was performed first
through an anterior orbitotomy via an upper lid
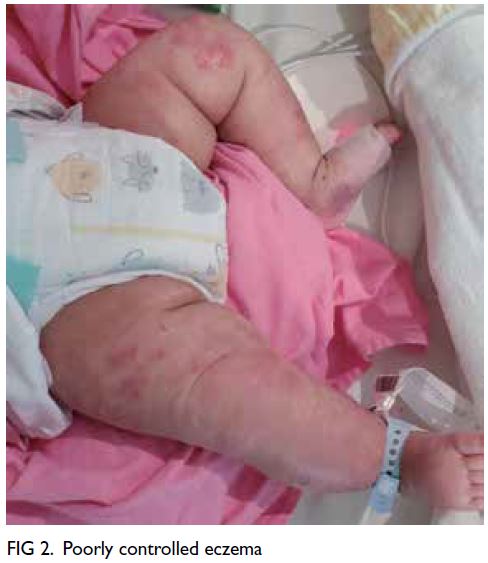
skin crease approach. Frozen section of the medial
yellow jelly-like mass revealed atypical lymphoid
cells with enlarged vesicular nuclei and amphophilic
cytoplasm, highly suspicious of lymphoproliferative
malignancy. We then performed a complete excision
of the vascular encapsulated intraconal lesion
using cryotherapy via a lateral orbitotomy. Formal
histopathology reports revealed the first medial
infiltrative lesion to be consistent with diffuse large
B-cell lymphoma with positive immunostaining for
CD20, BCL2, BCL6, MUM1, and CMYC1 (Fig 2).
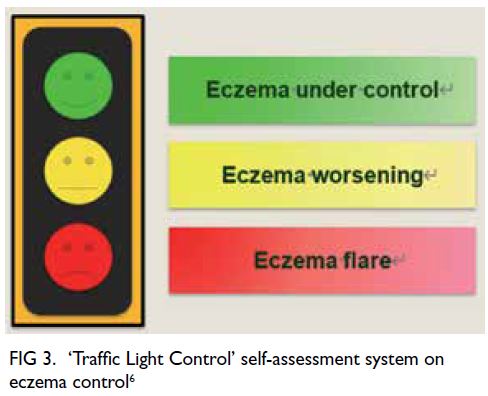
The second intraconal lesion was consistent with
cavernous haemangioma (Fig 3).
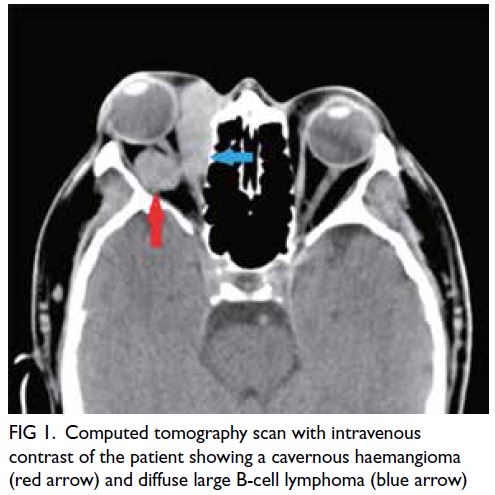
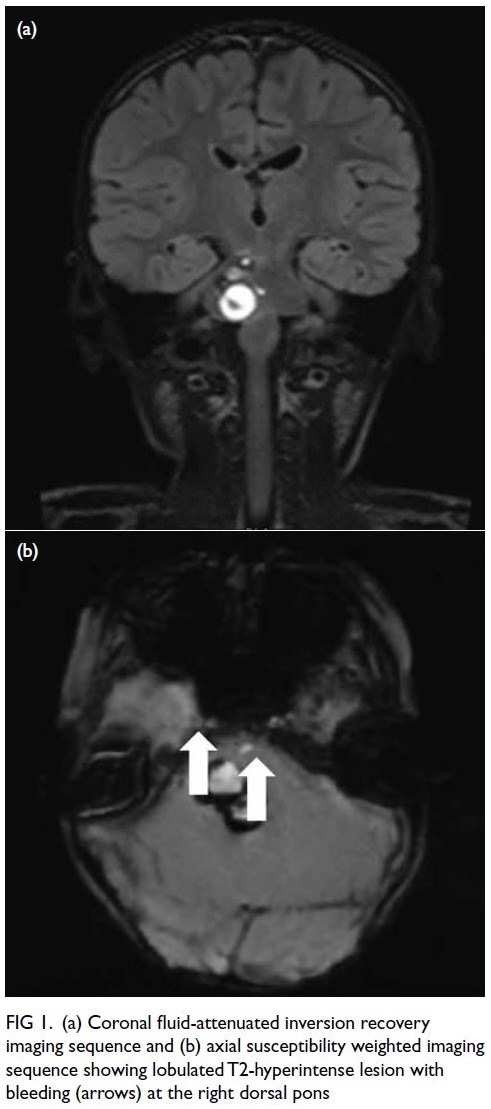
Figure 1. Computed tomography scan with intravenous contrast of the patient showing a cavernous haemangioma (red arrow) and diffuse large B-cell lymphoma (blue arrow)
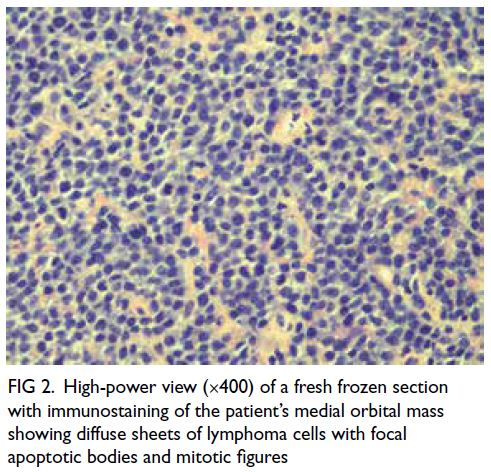
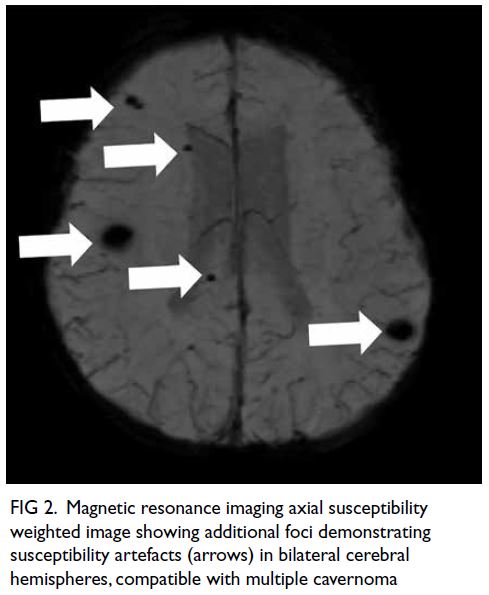
Figure 2. High-power view (×400) of a fresh frozen section with immunostaining of the patient’s medial orbital mass showing diffuse sheets of lymphoma cells with focal apoptotic bodies and mitotic figures
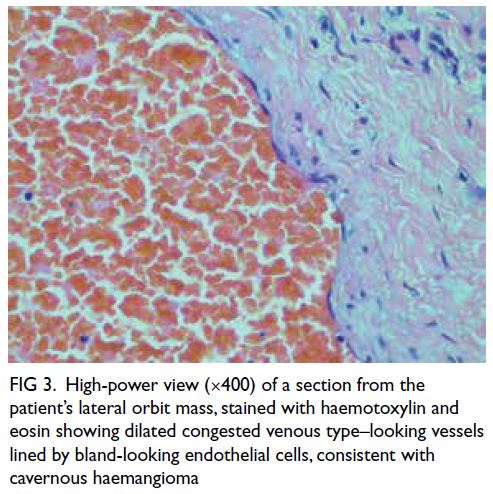
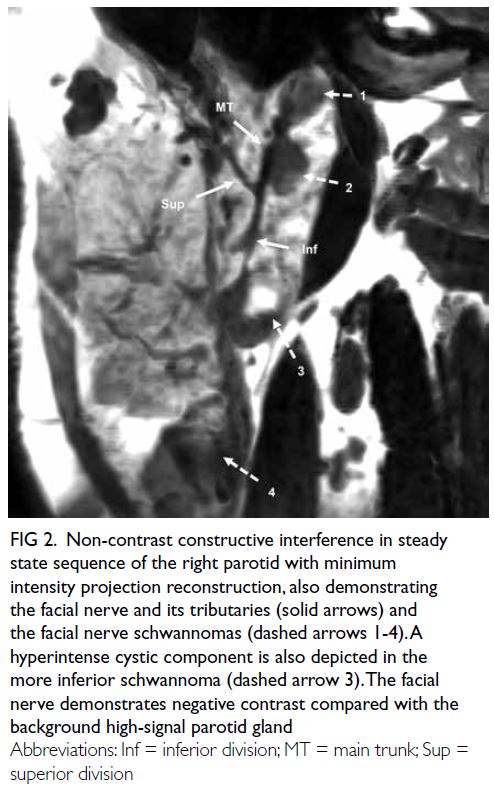
Figure 3. High-power view (×400) of a section from the patient’s lateral orbit mass, stained with haemotoxylin and eosin showing dilated congested venous type–looking vessels lined by bland-looking endothelial cells, consistent with cavernous haemangioma
At 4 months post-surgery there was no clinical
sign of optic nerve damage and best-corrected visual
acuity was 0.8 in the right eye. Positron emission
tomography–computed tomography showed a
residual hypermetabolic lesion over the medial aspect of the right orbit with no extra orbital lesion.
The patient is receiving chemotherapy under the
care of our haematology team.
Benign multiple homogenous lesions in
the same orbit have been reported. Although
multiple solitary fibrous tumours of the same
orbit without malignant degeneration have been
reported,4 multiple heterogeneous tumours in the
same orbit are extremely rare. Ma et al5 reported
a case of concurrent schwannoma and cavernous
haemangioma in the same orbit of a 54-year-old
female. To the best of our knowledge, concurrent
benign and malignant lesions of the same orbit have
not been reported in the English literature.
Author contributions
All authors contributed to the concept and design of this
study, acquisition of data, analysis of data, drafting the
manuscript, and critical revision for important intellectual content. All authors had full access to the data, contributed to
the study, approved the final version for publication, and take
responsibility for its accuracy and integrity.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration
of Helsinki and provided informed consent for the treatment
and consent for publication.
References
1. Demirci H, Shields CL, Shields JA, Honavar SG, Mercado GJ,
Tovilla JC. Orbital tumors in the older adult population.
Ophthalmology 2002;109:243-8. Crossref
2. Deng C, Hu W. Multiple cavernous hemangiomas in the
orbit: a case report and review of the literature. Medicine
(Baltimore) 2020;99:e20670. Crossref
3. Du B, He X, Wang Y, He W. Multiple recurrent
myxofibrosarcoma of the orbit: case report and review of
the literature. BMC Ophthalmol 2020;20:264. Crossref
4. Griepentrog GJ, Harris GJ, Zambrano EV. Multiply
recurrent solitary fibrous tumor of the orbit without
malignant degeneration: a 45-year clinicopathologic case
study. JAMA Ophthalmol 2013;131:265-7. Crossref
5. Ma M, Su F, Yang X. Multiple heterogeneous tumors in
orbit: a case report. Int J Clin Exp Pathol 2019;12:4137-41.
 A video clip demonstrating physical examination findings of bilateral fixed left lateral gaze and right lower-motor-neuron seventh cranial nerve palsy is available at
A video clip demonstrating physical examination findings of bilateral fixed left lateral gaze and right lower-motor-neuron seventh cranial nerve palsy is available at