Unilateral vocal cord palsy in a patient with jugular foramen schwannoma
Hong
Kong Med J 2021 Aug;27(4):303.e1–2
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Unilateral vocal cord palsy in a patient with
jugular foramen schwannoma
WK Wong, BDS, MD, CY Cheng, MD, WC Cheng, MD
Department of Neurosurgery, Chang Gung Memorial Hospital, Chiayi, Taiwan
Corresponding author: Dr WC Cheng (wancheng7511@yahoo.com.tw)
 Full
paper in PDF
Full
paper in PDF
A 74-year-old man presented with a history of
hoarseness of voice and choking for a few weeks.
Laryngoscopy revealed left vocal cord palsy (Fig 1).
Non-contrast computed tomography (CT) showed
no pulmonary or neck lesion, but one lobulated,
centrally low attenuating mass. T2-weighted
magnetic resonance images confirmed a 3.8-cm soft
tissue mass with heterogeneous enhancement in the
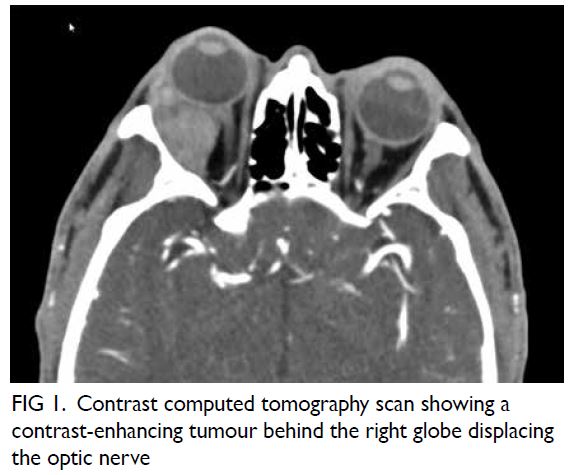
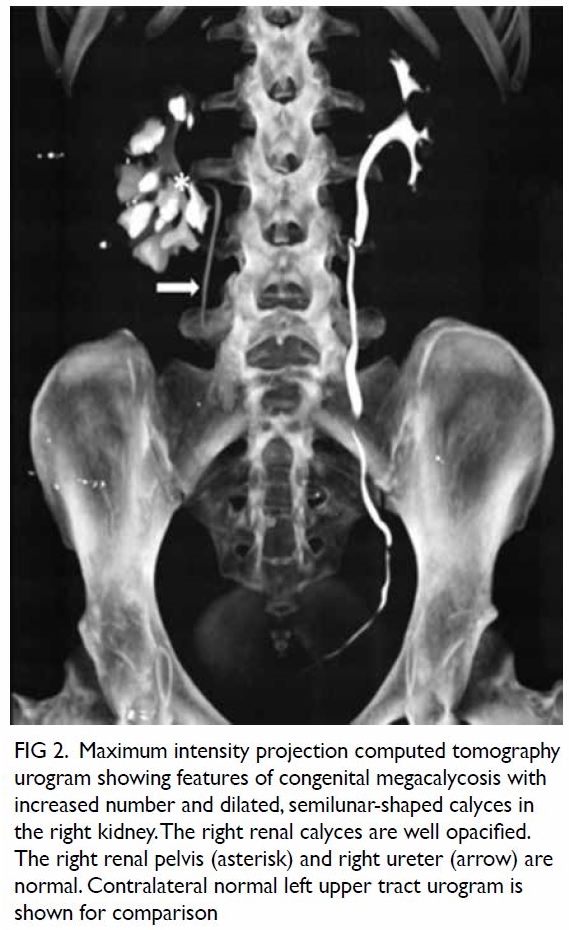
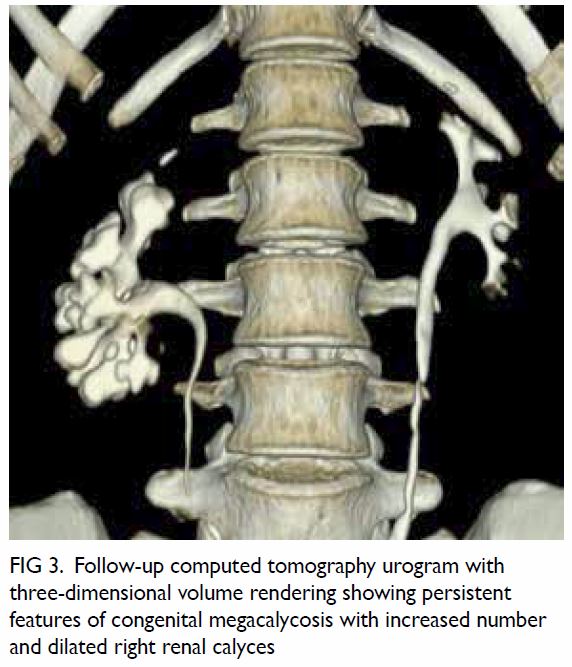
left cerebellopontine angle. In both images there was extension of the mass through an expansion of the
left jugular foramen, widened left internal acoustic
meatus and skull base destruction (Fig 2), compatible
with jugular foramen tumour. Subsequently the
patient developed progressive globus sensation,
tinnitus, left sternocleidomastoid muscle wasting,
and left neurosensory hearing loss. The tumour
was resected via a retrosigmoid approach. Surgical
assessment of the tumour revealed its origin from
cranial nerves IX and X and compression of cranial
nerves VIII and XI. The histopathological diagnosis
with immunohistochemistry staining was that of
schwannoma. Postoperative imaging demonstrated
adequate resection of the tumour. Improvement in
swallowing and hearing function test from severe to
moderate impairment were noted postoperatively.
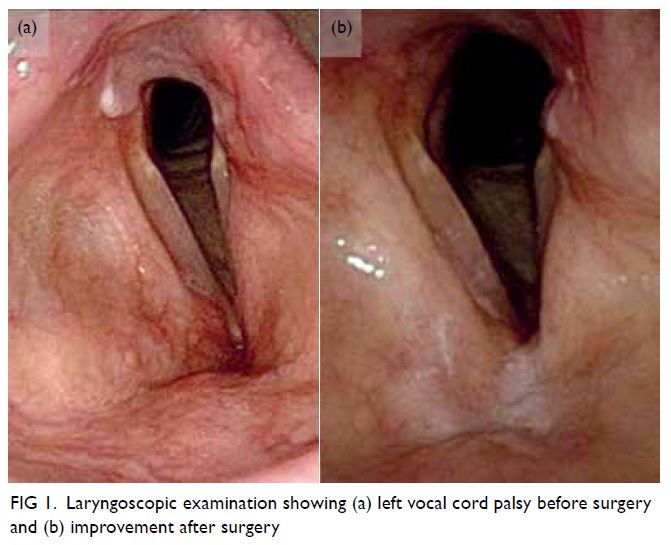
Figure 1. Laryngoscopic examination showing (a) left vocal cord palsy before surgery and (b) improvement after surgery
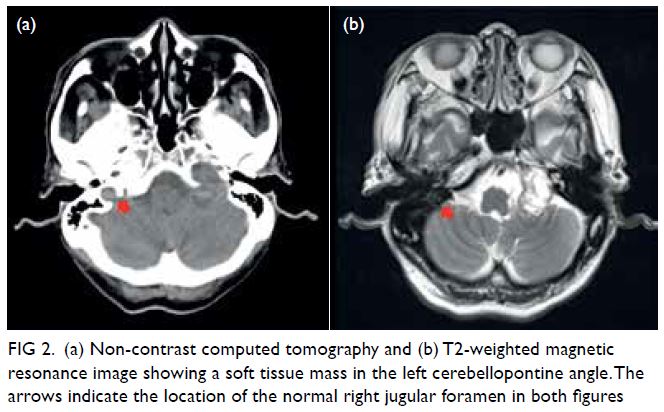
Figure 2. (a) Non-contrast computed tomography and (b) T2-weighted magnetic resonance image showing a soft tissue mass in the left cerebellopontine angle. The arrows indicate the location of the normal right jugular foramen in both figures
Discussion
Schwannoma is a benign tumour of the nerve sheath.
The most common intracranial schwannoma is
vestibular schwannoma (acoustic neuroma) that
arises from cranial nerve VIII.1 Jugular foramen
schwannomas, which mainly arise from cranial nerves
IX and X, account for around 3% to 4% of all intracranial
schwannomas. They are more prevalent in women
and occur between the third and sixth decades of life.
Clinical presentation is variable because of their slow-growing
nature and proximity to other cranial nerves.
Symptoms appear when the tumour is sufficiently
large and most commonly consist of hearing loss,
tinnitus, dysphagia, ataxia, and hoarseness. Other
symptoms include dysarthria, dysphonia, aspiration,
vertigo, dizziness, shoulder weakness, and headache.2
Differential diagnoses of unilateral vocal cord palsy
are usually divided into malignancies of the lung and
the neck and non-malignant lesions of traumatic,
neurological, inflammatory or infectious origin. A
CT scan is indicated to evaluate the more common
pulmonary malignancies and to ensure presence of a
rarer lesion along the course of the recurrent laryngeal
and vagus nerve up to the skull base is not missed,
especially if signs and symptoms of multiple cranial
nerve involvement are present.3
Imaging studies can help differentiate vestibular
and lower cranial nerve schwannomas, meningioma,
glomus jugulare paraganglioma, ependymomas,
and metastatic tumour. Imaging findings of jugular
foramen schwannoma include scalloped and sclerotic
expansion of the temporal bone instead of a lytic pattern. On magnetic resonance imaging, the lesion
is T1 iso- or hypo-intense or T2 iso- or hyper-intense
with gadolinium enhancement, whereas on CT, it is
isodense to brain parenchyma and enhanced with
contrast.4 The site of origin is classified as cisternal,
foraminal (intraosseous) or extracranial. However, it
is difficult to ascertain the exact site radiologically and
clinically because of the variable location and nerve
involvement. Assessment of the nerve root during
surgery is required to confirm the origin of the tumour.
The selection of surgical approach for treatment is
determined by the location, pattern and extension of
the tumour, the degree of bone destruction/erosion
and the neurological and preoperative hearing status.5
Risks of operation include damage to other cranial
nerves, especially facial nerve palsy, and incomplete
removal of the tumour leading to recurrence.
Author contributions
Concept or design: All authors.
Acquisition of data: WK Wong, CY Cheng.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: WK Wong.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: WK Wong, CY Cheng.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: WK Wong.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of Helsinki and provided consent for all investigations and
procedures.
References
1. Suri A, Bansal S, Singh M, Mahapatra AK, Sharma BS. Jugular foramen schwannomas: a single institution patient
series. J Clin Neurosci 2014;21:73-7. Crossref
2. Bakar B. The jugular foramen schwannomas: review of the large surgical series. J Korean Neurosurg Soc 2008;44:285-94. Crossref
3. Stimpson P, Patel R, Vaz F, et al. Imaging strategies for investigating unilateral vocal cord palsy: how we do it. Clin Otolaryngol 2011;36:266-71. Crossref
4. Lee M, Tong K. Jugular foramen schwannoma mimicking paraganglioma: case report and review of imaging
findings. Radiol Case Rep 2016;11:25-8. Crossref
5. Samii M, Alimohamadi M, Gerganov V. Surgical treatment
of jugular foramen schwannoma: surgical treatment based
on a new classification. Neurosurgery 2015;77:424-32. Crossref