Hong
Kong Med J 2021 Jun;27(3):223.e1–2
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Primary orbital melanoma
Vivian WK Hui, MB, ChB1; TC Lau, PhD2; Lawrence PW Ng, MSc2; Hunter KL Yuen, FRCOphth1; W Cheuk, FHKCPath2
1 Department of Ophthalmology, Hong Kong Eye Hospital, Hong Kong
2 Department of Pathology, Queen Elizabeth Hospital, Hong Kong
Corresponding author: Dr W Cheuk (cwzz01@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
A 68-year-old Chinese man presented with proptosis,
epiphora and discomfort in the right eye associated
with impaired visual acuity. Contrast computed
tomography scan revealed a 3.3 cm × 2.1 cm
heterogeneously enhancing mass at the
superotemporal aspect of the right orbit with
displacement of the lateral rectus muscle (Fig 1).
A pigmented tumour with dense adhesions to the
surrounding structures was found in orbitotomy
with leakage of the pigmented content upon surgical
exploration. The lesion was excised as much as
possible. Detailed clinical examination showed no
evidence of intraocular melanoma, conjunctival
or eyelid melanoma, or melanoma anywhere else.
Systemic examination and whole-body positron
emission tomography-computed tomography scan
were unremarkable. The patient was alive with no
evidence of disease at 9-month follow-up.
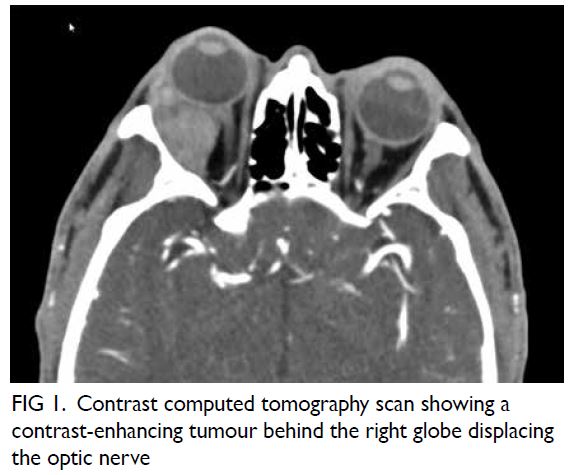
Figure 1. Contrast computed tomography scan showing a contrast-enhancing tumour behind the right globe displacing the optic nerve
The tumour was a solid, dark brownish
multinodular mass covered by fat and skeletal
muscle, comprising lobulated sheets of epithelioid
and spindly melanocytes with vesicular chromatin,
prominent nucleoli, and variable amounts of
melanin pigment (Figs 2, 3, and 4). The mitotic count
was 1 per 10 HPFs. The neoplastic melanocytes were
positive for S100 and SOX10 but negative for BRAF.
BRCA1-associated protein 1 immunostaining was
intact. Sanger sequencing revealed a GNA11 Q209L
mutation. No mutation was found in GNAQ, NRAS,
KRAS, BRAF or KIT.
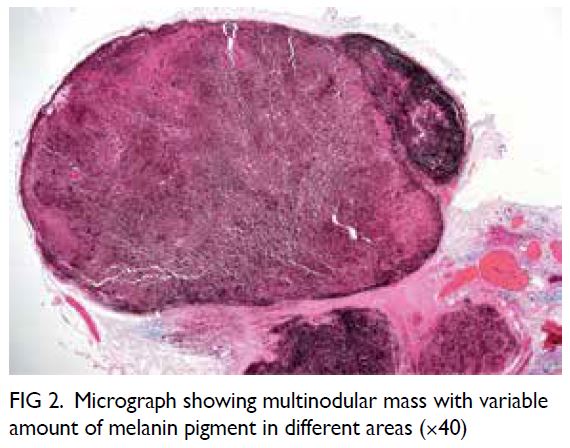
Figure 2. Micrograph showing multinodular mass with variable amount of melanin pigment in different areas (×40)
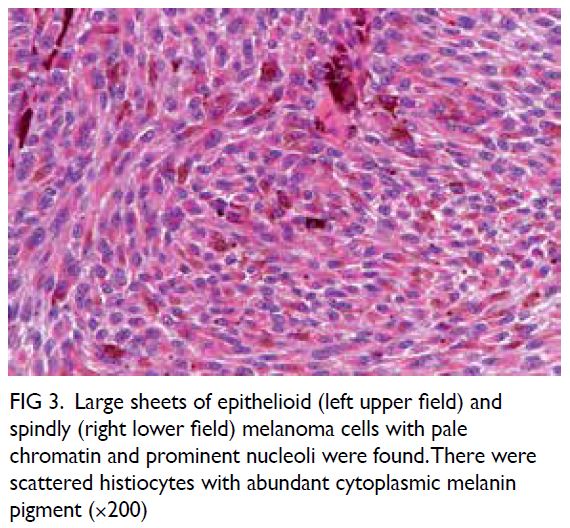
Figure 3. Large sheets of epithelioid (left upper field) and spindly (right lower field) melanoma cells with pale chromatin and prominent nucleoli were found. There were scattered histiocytes with abundant cytoplasmic melanin pigment (×200)
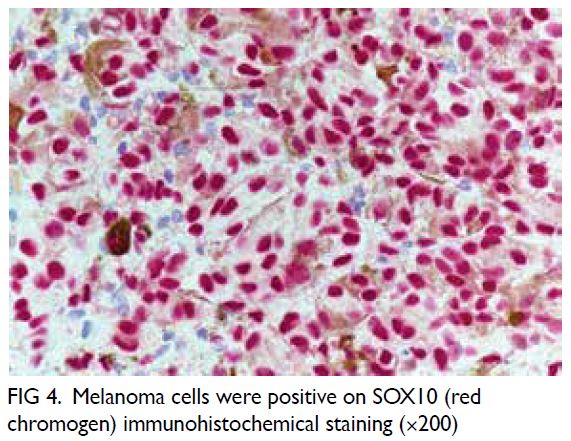
Figure 4. Melanoma cells were positive on SOX10 (red chromogen) immunohistochemical staining (×200)
Ocular melanoma most frequently involves
the uveal tract (choroid, ciliary body, and iris) and
the conjunctiva, where melanocytes are normally present. According to The Cancer Genome Atlas
uveal melanoma project, mutually exclusive
mutations in GNAQ, GNA11, CYSLTR2 and PLCB4
are the tumour-initiating mutations whereas BAP1,
EIF1AX and SF3B1/SRSF2 mutations are associated
with prognosis in uveal melanoma.1 In contrast,
conjunctival melanoma typically exhibits BRAF,
NRAS and NF1 mutations of the MAPK pathway
with UV-induced C→T mutation signature.2
Orbital soft tissue melanoma is uncommon with
>90% representing secondary tumours as a result
of contiguous spread or metastasis from uveal,
conjunctival, cutaneous or sinonasal melanomas.3
Primary orbital melanoma is exceedingly rare
and has been postulated to arise in embryological
remnants of melanocytes arrested in the region since
a substantial proportion of cases are associated with
blue nevus, orbital melanocytosis or oculodermal
melanocytosis (nevus of Ota). Alternatively, it may
arise from isolated melanocytes found in the optic
nerve sheath, orbital fat, extraocular muscles or
orbital periosteum. The majority of patients are
white Europeans in their fourth to fifth decade.
Histologically, it comprises epithelioid, spindle or
mixed populations of neoplastic melanocytes that
are frequently pigmented but with a lesser degree
of nuclear pleomorphism than their cutaneous and
mucosal counterparts. The prognosis is generally
poor but a small proportion of patients survive
long term. The most frequent site of metastasis
is the liver.3 4 The clinicopathological features of
primary orbital melanoma are very similar to those
of uveal melanoma. The findings of GNAQ, SF3B1,
EIF1AX mutations5 and GNA11 mutation in this
case provide further evidence that primary orbital
melanoma has a close pathogenic relationship with
uveal melanoma.
Author contributions
All authors contributed to the concept or design of the study, acquisition of the data, analysis or interpretation of the
data, drafting of the manuscript, and critical revision of the
manuscript for important intellectual content. All authors
had full access to the data, contributed to the study, approved
the final version for publication, and take responsibility for its
accuracy and integrity.
Conflicts of interest
As an adviser of the journal, HKL Yuen was not involved in the peer review process. Other authors have disclosed no
conflicts of interest.
Declaration
This case was presented as a poster at the Annual Scientific Meeting of The College of Ophthalmologists of Hong Kong
and Hong Kong Ophthalmological Society on 14 to 15
December 2019.
Funding/support
This pictorial medicine paper received no specific grant from any funding agency in the public, commercial, or not-forprofit
sectors.
Ethics approval
The patient was treated in accordance with the tenets of the Declaration of Helsinki. Informed consent was obtained from
the patient.
References
1. Bakhoum MF, Esmaeli B. Molecular characteristics of uveal melanoma: insights from the Cancer Genome Atlas
(TCGA) Project. Cancers (Basel) 2019;11:1061.Crossref
2. Swaminathan SS, Field MG, Sant D, et al. molecular characteristics of conjunctival melanoma using whole-exome
sequencing. JAMA Ophthalmol 2017;135:1434-7. Crossref
3. Rose AM, Luthert PJ, Jayasena CN, Verity DH, Rose GE. Primary orbital melanoma: presentation, treatment,
and long-term outcomes for 13 patients. Front Oncol
2017;7:316. Crossref
4. Shields JA, Shields CL. Orbital malignant melanoma: the 2002 Sean B Murphy lecture. Ophthalmic Plast Reconstr
Surg 2003;19:262-9. Crossref
5. Rose AM, Luo R, Radia UK, et al. Detection of mutations in SF3B1, EIF1AX and GNAQ in primary orbital melanoma
by candidate gene analysis. BMC Cancer 2018;18:1262. Crossref