Use of colposcopy in a patient with recurrent genital ulcers
Hong Kong Med J 2015 Aug;21(4):375.e3–4
DOI: 10.12809/hkmj154536
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Use of colposcopy in a patient with recurrent genital ulcers
Tomoko Matsuzono, MRCOG, FHKAM (Obstetrics and Gynaecology);
WH Li, FHKCOG, FHKAM (Obstetrics and Gynaecology)
Department of Obstetrics and Gynaecology, Queen Elizabeth Hospital, Jordan, Hong Kong
Corresponding author: Dr Tomoko Matsuzono (tomoko821@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Case
The patient was a 40-year-old female with a 5-month
history of recurrent painful labial ulcers in December
2013. Investigations by a private physician revealed
a negative VDRL (Venereal Disease Research
Laboratory) test and vulval swab culture grew
commensals only. The ulcers, however, failed to heal
following treatment with antibiotics.
Vulval biopsy was performed at her first
visit and histopathology report revealed only a
non-specific ulcer with negative stains for fungus,
bacteria, acid-fast bacilli, and herpes simplex virus.
Papanicolaou smear showed atypical squamous cells
of unknown significance (ASCUS).
In the absence of any obvious pathology for
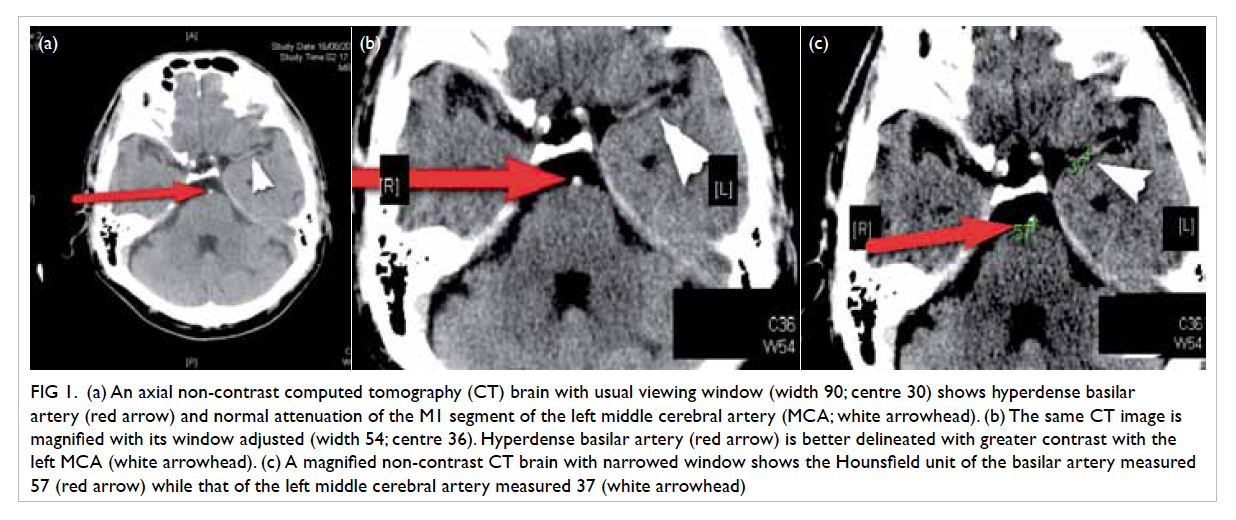
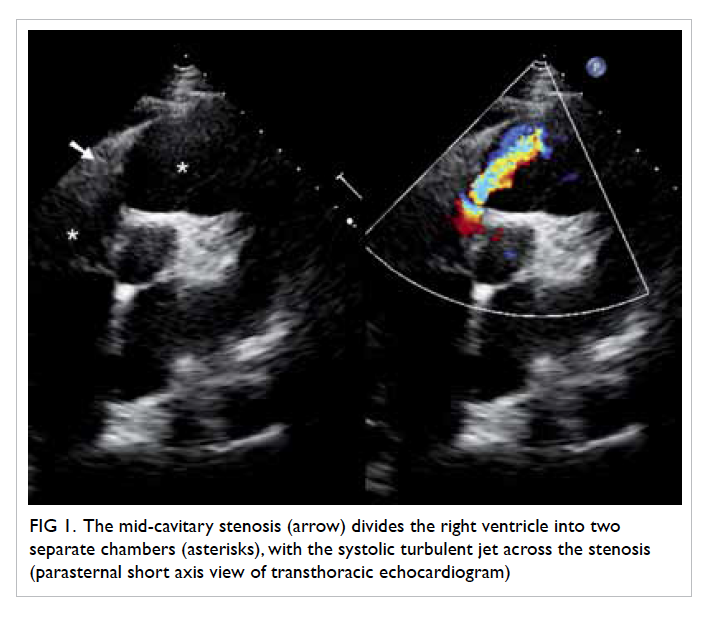
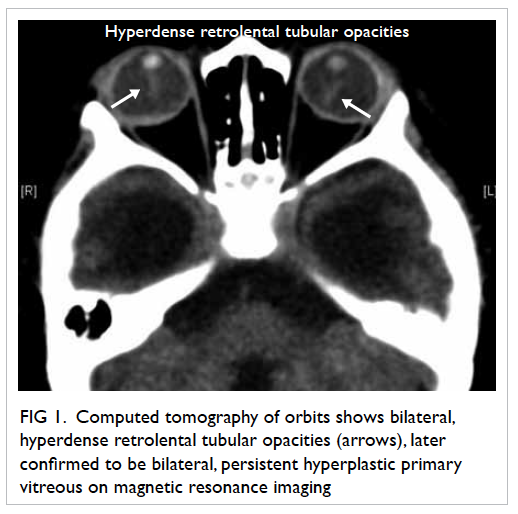
the non-healing ulcer, colposcopy was arranged: four
ulcers were identified within the vagina, scattered
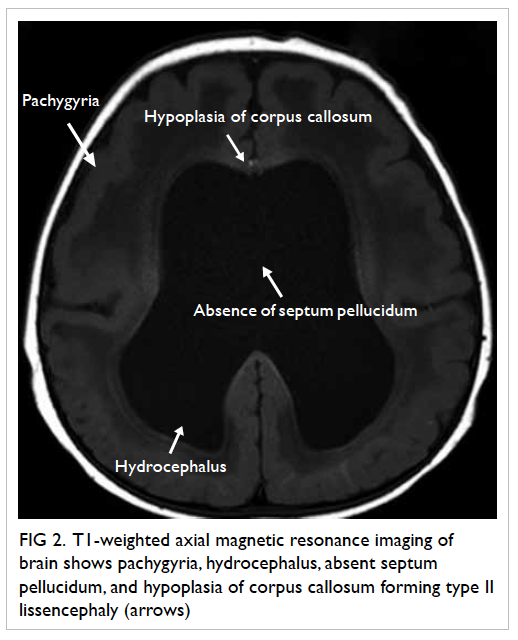
over the right and left vaginal walls (Fig 1). A left lower
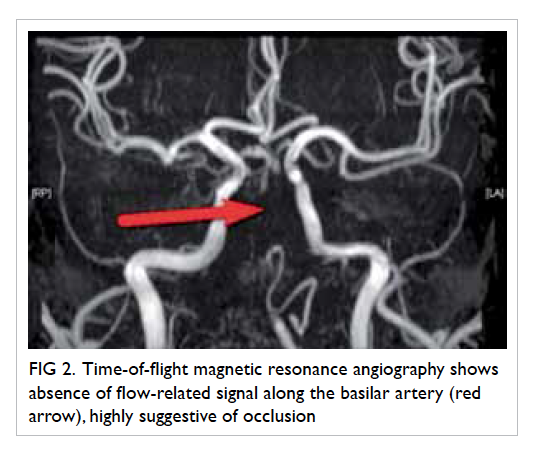
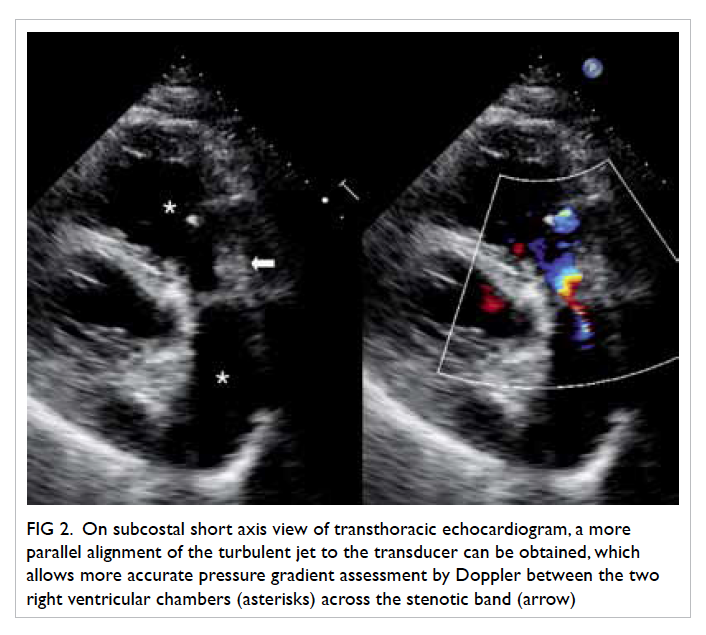
vulval ulcer measuring 3 cm with slightly raised
edges was also seen (Fig 2). Biopsies were taken over
the cervix, vaginal, and vulval ulcers. Histopathology
of these biopsies confirmed the presence of cervicitis
and ulcers, with no evidence of malignancy.
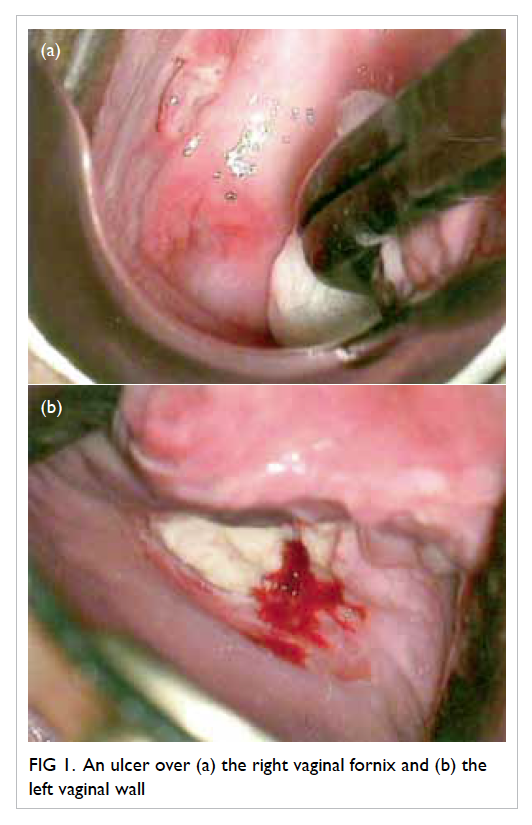
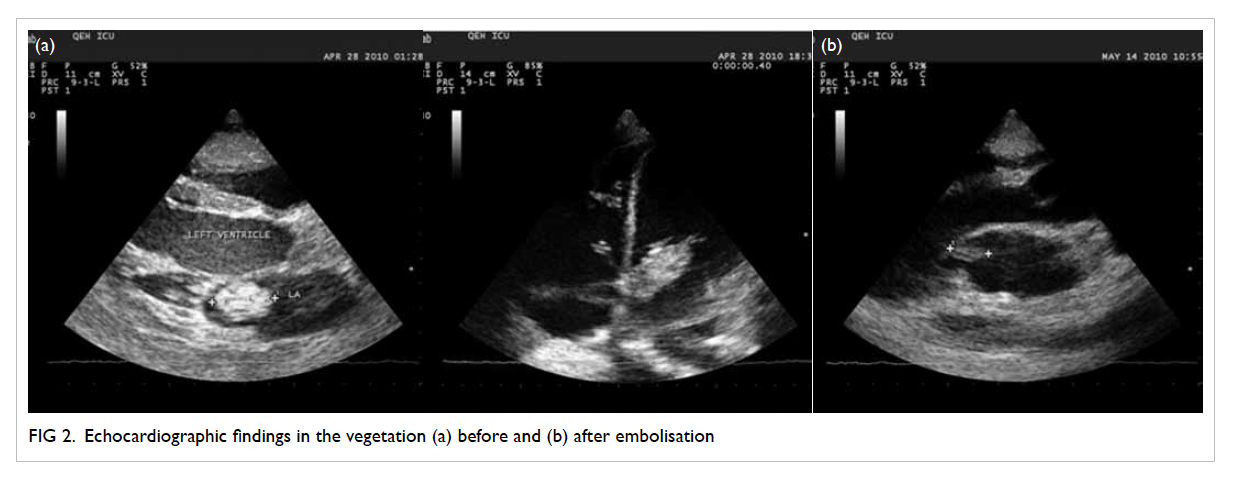
Figure 1. An ulcer over (a) the right vaginal fornix and (b) the left vaginal wall
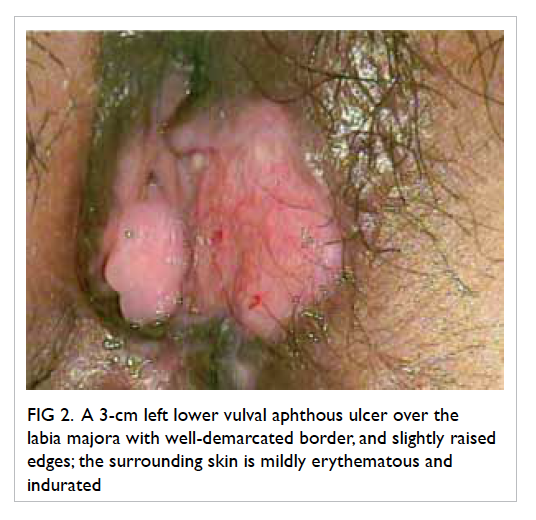
Figure 2. A 3-cm left lower vulval aphthous ulcer over the labia majora with well-demarcated border, and slightly raised edges; the surrounding skin is mildly erythematous and indurated
Upon systemic review, the patient revealed
that she had been suffering from recurrent mouth
ulcers and bruising over both shins. Subsequent
examination revealed multiple aphthous mouth
ulcers, as well as erythema nodosum over both shins.
Based on the clinical signs, a diagnosis
of Behçet’s disease was made. The patient was
commenced on prednisolone, and all ulcers had
healed at subsequent follow-up.
Discussion
Behçet’s disease is a rare cause of genital ulceration. It
is a multisystem vasculitic disorder and the diagnosis
is based on clinical criteria.1 2 3 4 Our patient fulfilled
the criteria of recurrent oral and genital ulcerations
as well as a cutaneous manifestation.
Although there is little published evidence
to support the value of colposcopy and biopsy
in the diagnosis of Behçet’s disease, colposcopy
enables a more detailed evaluation of the genital
tract, and can be used to exclude the possibility of
malignancy. Histopathological results in Behçet’s
ulcer are typically non-specific with chronic active inflammation.
Papanicolaou smear in our patient showed
ASCUS, but histopathology result for the cervix was
normal. In a prospective study by Özdemir et al,2
abnormal cervical cytology, acetowhite epithelium,
and iodine-negative epithelium on colposcopy were
more common in Behçet’s patients. Nonetheless, the
majority of ASCUS results revealed a normal finding
for cervical histopathology. It was suggested that this
was due to the benign inflammatory changes in the
cervical epithelium.2
As in this case, diagnosis for recurrent genital
ulcers can be difficult although when information
about oral ulcers became available, a diagnosis of
Behçet’s became more obvious. A complete history
and physical examination that pays particular
attention to signs or symptoms of an underlying
associated systemic condition are essential when
evaluating patients with recurrent genital ulcers.
References
1. Patel S, Prime K. Recurrent vulval ulceration: could it be
Behçet’s disease? Int J STD AIDS 2012;23:683-4. Crossref
2. Özdemir S, Özdemir M, Celik C, Balevi A, Toy H, Kamış
U. Evaluation of patients with Behçet’s disease by cervical
cytology and colposcopic examination. Arch Gynecol
Obstet 2012;285:1363-8. Crossref
3. Keogan MT. Clinical Immunology Review Series: an
approach to the patient with recurrent orogenital
ulceration, including Behçet’s syndrome. Clin Exp
Immunol 2009;156:1-11. Crossref
4. Bandow GD. Diagnosis and management of vulvar ulcers.
Dermatol Clin 2010;28:753-63. Crossref