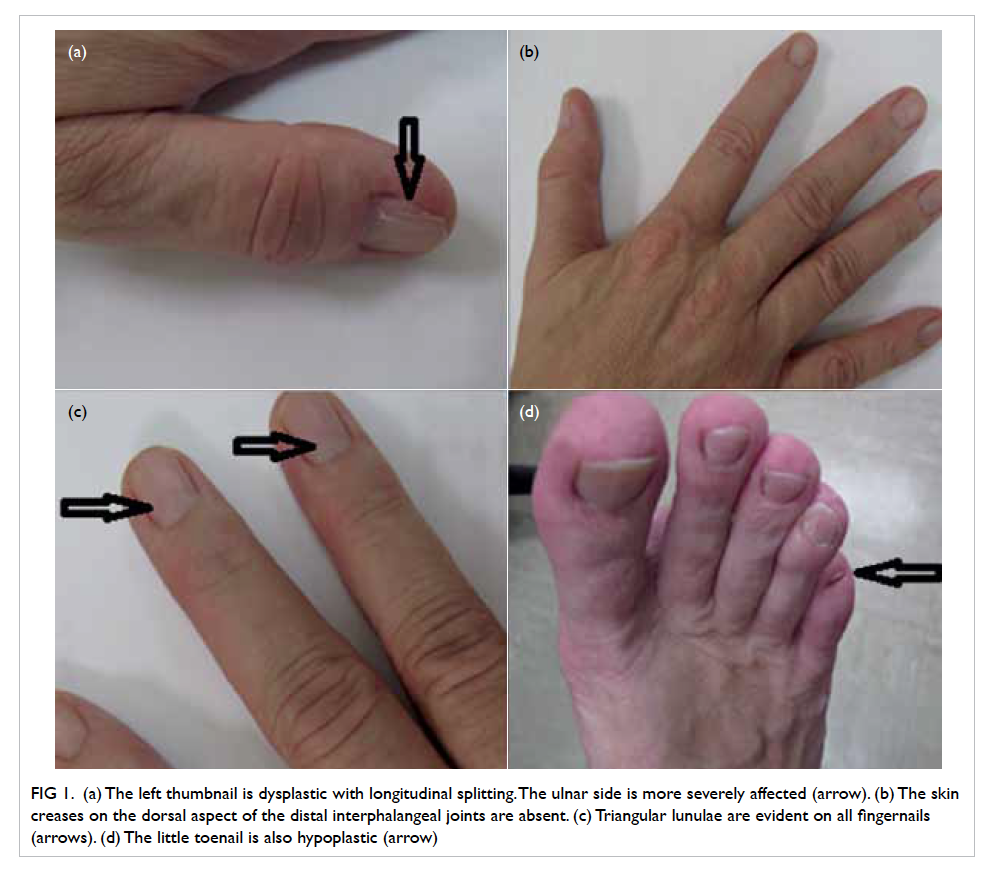
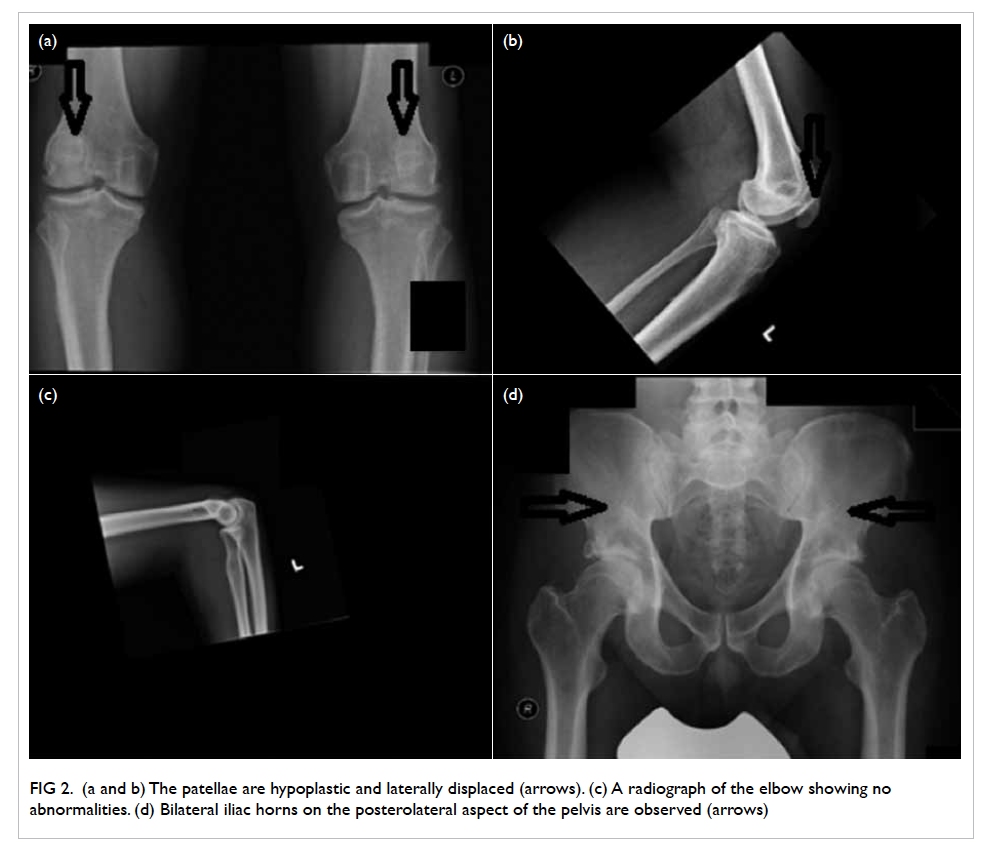
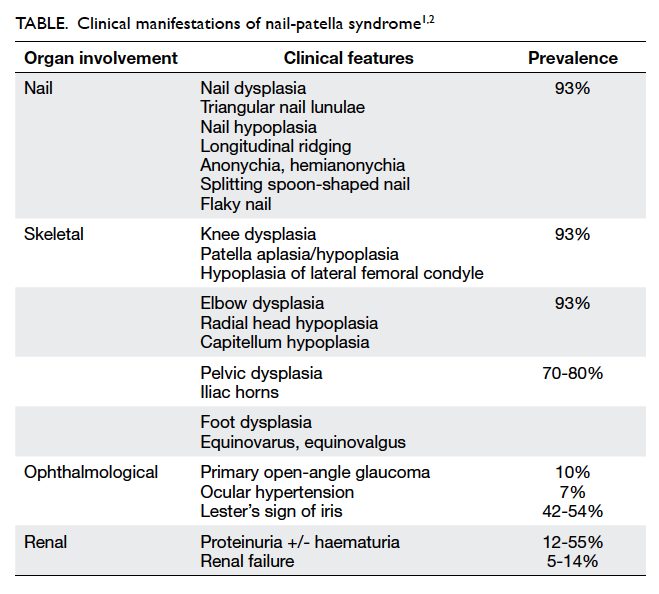
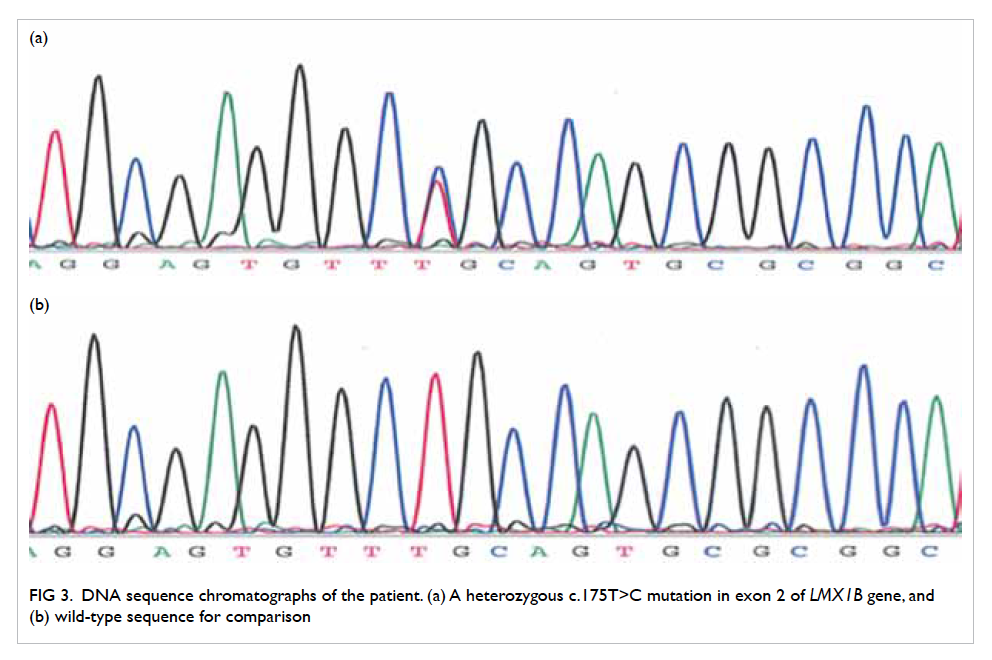
Virilisation in a menopausal woman with a previous kidney transplant
Hong Kong Med J 2016 Dec;22(6):623.e3–4
DOI: 10.12809/hkmj164901
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Virilisation in a menopausal woman with a previous kidney transplant
TM Fung, FRCOG, FHKAM (Obstetrics and Gynaecology)1;
WC Wong, FHKCPath, FHKAM (Pathology)2;
KW Chan, FHKCP, FHKAM (Medicine)3;
KS Fung, FHKCP, FHKAM (Medicine)3
1 Department of Obstetrics and Gynaecology, Princess Margaret Hospital, Laichikok, Hong Kong
2 Department of Clinical Pathology, Pamela Youde Nethersole Eastern
Hospital, Chai Wan, Hong Kong
3 Department of Medicine and Geriatrics, Princess Margaret Hospital,
Laichikok, Hong Kong
Corresponding author: Dr TM Fung (fungtm@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
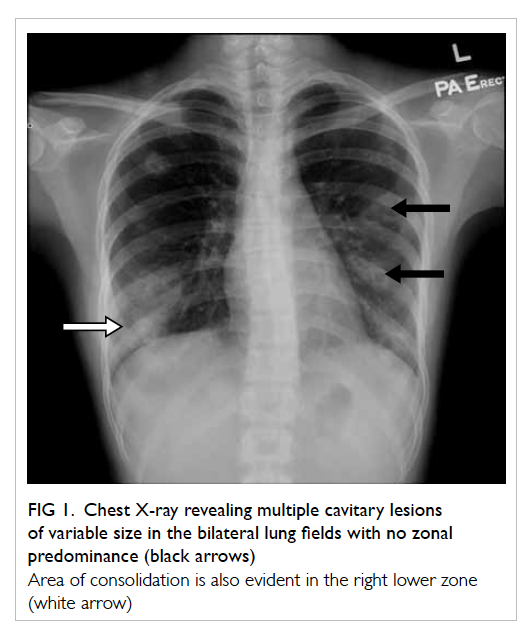
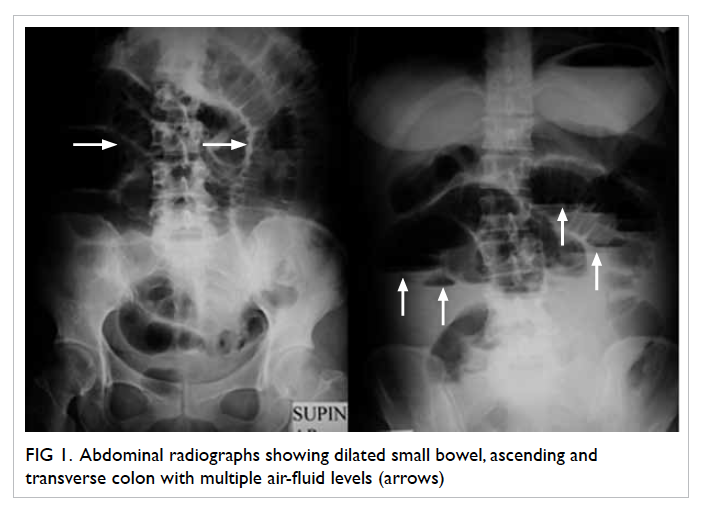
A 53-year-old woman, menopausal for 4 years and
with a previous kidney transplant presented with a
3-year history of excessive facial and body hair that
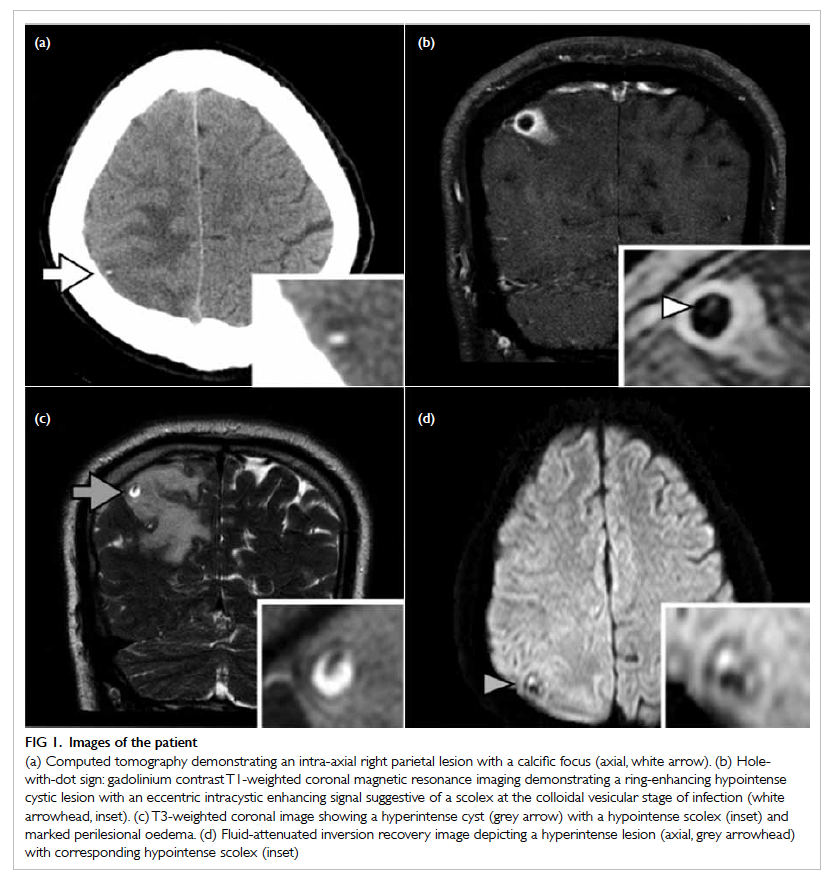
required daily shaving in January 2008 (Fig 1). She had
had no postmenopausal bleeding or gynaecological
disease. She was prescribed immunosuppressants
and corticosteroids but no hormones. There was no
acne or hoarseness of voice. Examination revealed
frontal balding and excessive hair over her chin, both
shins, and lower abdomen with mild clitoromegaly.
Serum testosterone was 19 nmol/L (reference range,
0.35-2.65 nmol/L); 17-hydroxyprogesterone (17-OHP) and dehydroepiandrosterone sulfate (DHEAS)
levels were normal. Adrenocorticotropic hormone
and cortisol levels were normal after overnight
dexamethasone suppression test. Her follicle-stimulating
hormone, luteinising hormone, thyroid-stimulating
hormone, and prolactin levels were also
normal. Serum oestradiol was in the premenopausal
range (298 nmol/L). Transvaginal ultrasound showed
a normal uterus with endometrial thickness of 5 mm
and no adnexal masses.
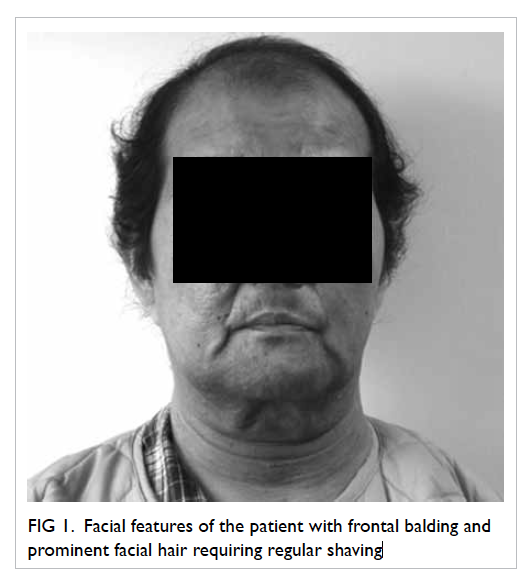
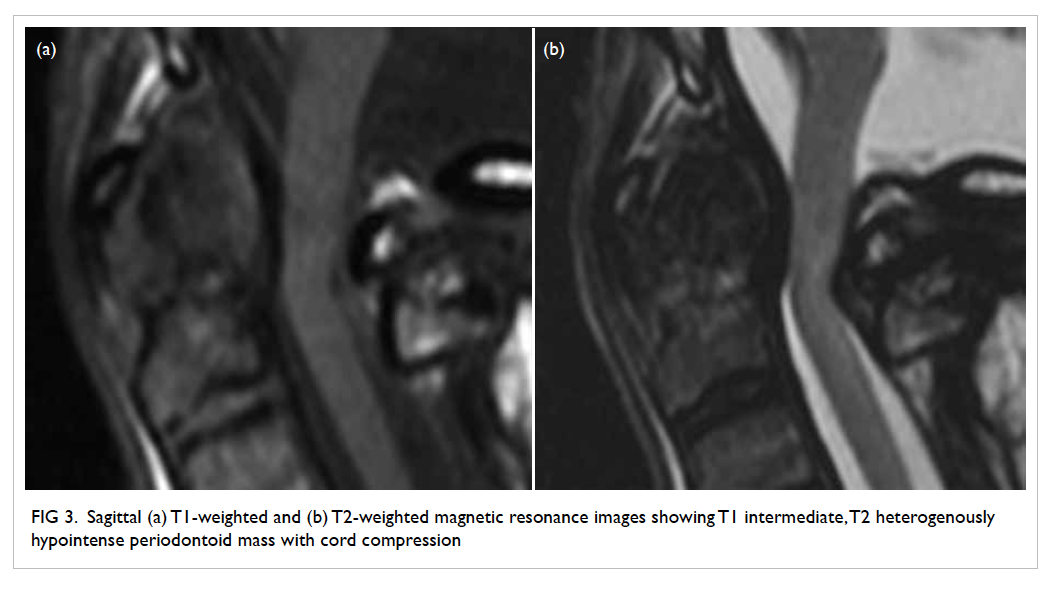
Figure 1. Facial features of the patient with frontal balding and prominent facial hair requiring regular shaving
Questions:
1. What are the differential diagnoses?
2. What further investigations are helpful?
3. How should the patient be managed?
Answers:
1. Androgen-secreting tumours of the ovary or
adrenal gland.
Hirsutism involves excessive terminal hair growth in a masculine distribution in women. It can occur as a side-effect of immunosuppressants, eg cyclosporin with gingival hyperplasia and hirsutism. Nonetheless virilisation is uncommon. Cyclosporin should be changed to tacrolimus when hirsutism occurs.
Virilisation (development of male characteristics in women) occurs in less than 1% of patients with hirsutism. When it occurs (temporal hair recession and clitoromegaly in this patient) androgen-secreting tumours of the ovaries or adrenals should be suspected, particularly when onset is sudden with rapid progression.
Hirsutism involves excessive terminal hair growth in a masculine distribution in women. It can occur as a side-effect of immunosuppressants, eg cyclosporin with gingival hyperplasia and hirsutism. Nonetheless virilisation is uncommon. Cyclosporin should be changed to tacrolimus when hirsutism occurs.
Virilisation (development of male characteristics in women) occurs in less than 1% of patients with hirsutism. When it occurs (temporal hair recession and clitoromegaly in this patient) androgen-secreting tumours of the ovaries or adrenals should be suspected, particularly when onset is sudden with rapid progression.
2. Patients with virilisation should have serum
testosterone, 17-OHP, and DHEAS checked.
Serum testosterone level of >200 ng/dL (ie 6.94
nmol/L or 3 times normal) and/or DHEAS level
of >700 µg/dL (ie 24.3 nmol/L) are indicative of
virilising tumours.1 2 Computed tomography (CT)
of the adrenals and pelvis should be considered.
Ultrasonography of the pelvis may be difficult and
not diagnostic in menopausal women because the
ovaries are small and steroid-secreting tumours
can be <1 cm in diameter.3 In this patient, normal
levels of DHEAS and 17-OHP made adrenal
tumour and congenital adrenal hyperplasia
unlikely. Ovarian virilising tumours have to be
suspected. Preoperative CT pelvis showed a 1-cm
cyst in her right ovary.
3. Endometrial aspirate was performed, there was
no hyperplasia or malignancy. She underwent
laparotomy and bilateral salpingo-oophorectomy.
There was a 1-cm yellowish tumour in the
right ovary. The left ovary appeared normal.
Hysterectomy was not done as the uterus
was densely adhered to the bowel (history of
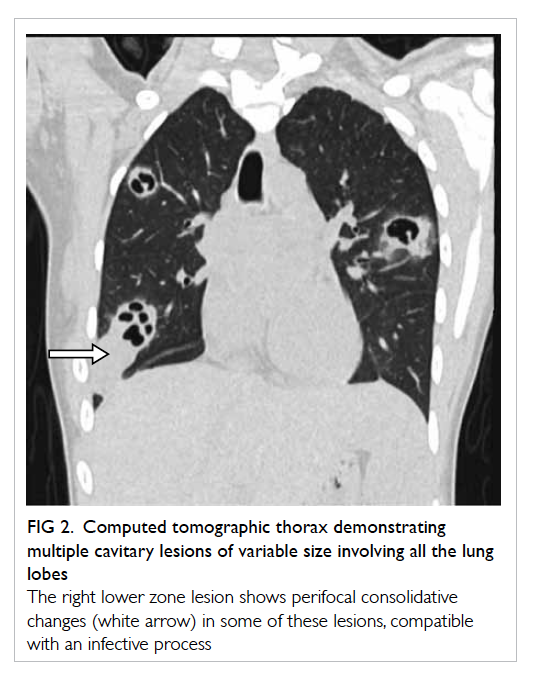
peritoneal dialysis). Histology confirmed stromal
luteoma and hyperthecosis in both ovaries
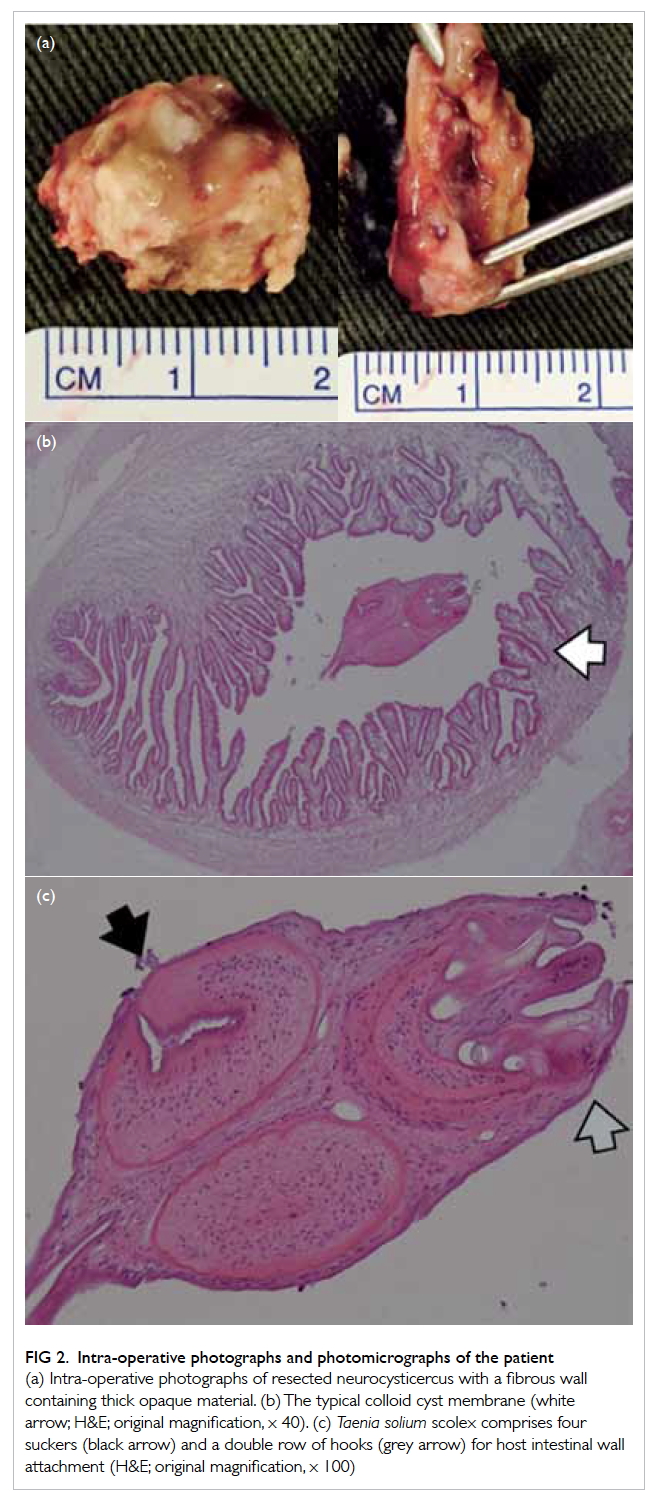
without malignancy (Fig 2). After surgery, her testosterone level normalised. Hair re-grew over
her frontal region and her body hair reduced.
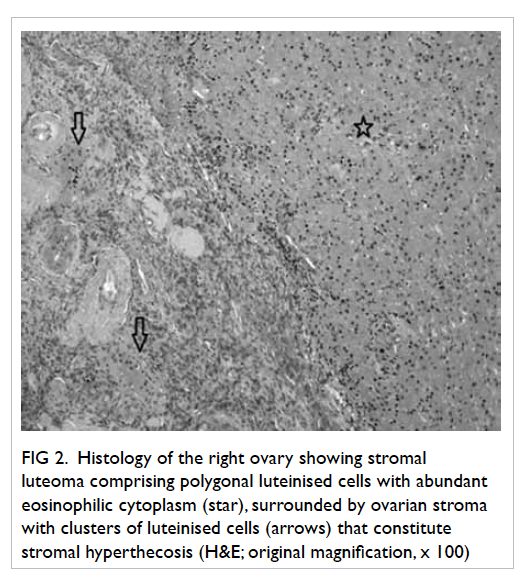
Figure 2. Histology of the right ovary showing stromal luteoma comprising polygonal luteinised cells with abundant eosinophilic cytoplasm (star), surrounded by ovarian stroma with clusters of luteinised cells (arrows) that constitute stromal hyperthecosis (H&E; original magnification, x 100)
Ovarian steroid cell tumours account for 0.1%
to 0.2% of all ovarian tumours, and are composed of
cells resembling steroid-secreting cells.4 Steroid cell
tumours of small size and confined to the ovarian
stroma are conventionally designated stromal
luteomas and are usually associated with stromal
hyperthecosis in adjacent stroma. They can have
oestrogenic and/or androgenic manifestations
with postmenopausal bleeding or virilisation, and
are associated with endometrial hyperplasia or
carcinoma.5 Hysterectomy and bilateral salpingo-oophorectomy is advised as some of these tumours
may have malignant potential.
References
1. Somani N, Harrison S, Bergfeld WF. The clinical evaluation
of hirsutism. Dermatol Ther 2008;21:376-91. Crossref
2. Hunter MH, Carek PJ. Evaluation and treatment of women
with hirsutism. Am Fam Physician 2003;67:2565-72.
3. Outwater EK, Wagner BJ, Mannion C, McLarney JK, Kim
B. Sex cord–stromal and steroid cell tumors of the ovary.
Radiographics 1998;18:1523-46. Crossref
4. Hayes MC, Scully RE. Stromal luteoma of the ovary: a
clinicopathological analysis of 25 cases. Int J Gynecol
Pathol 1987;6:313-21. Crossref
5. Yamada S, Tanimoto A, Wang KY, Shimajiri S, Sasaguri
Y. Stromal luteoma and nodular hyperthecosis of the
bilateral ovaries associated with atypical endometrial
hyperplasia of the uterus. Pathol Int 2009;59:831-3. Crossref