Successful endovascular treatment in a COVID-19 patient with mycotic aortoiliac aneurysm due to Salmonella typhi: a case report
Hong Kong Med J 2024 Feb;30(1):72–4 | Epub 8 Feb 2024
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Successful endovascular treatment in a COVID-19 patient with mycotic aortoiliac aneurysm due to Salmonella typhi: a case report
Samuel Edrei So, MB, BS; YC Chan, MB, BS, FRCS; Stephen W Cheng, MB, BS, FRCS
Division of Vascular Surgery, Department of Surgery, Queen Mary Hospital, Hong Kong SAR, China
Corresponding author: Dr YC Chan (ycchan88@hku.hk)
 Full paper in PDF
Full paper in PDF
Introduction
Conventional surgical options for mycotic aneurysm
include ligation or excision with in situ or extra-anatomical
reconstruction. Endovascular stenting in
the presence of sepsis is controversial but may be the
preferred management for critically ill patients who
are deemed very high risk for open intervention.1
Mycotic aneurysms due to Salmonella typhi are
extremely rare, with only two cases described in the
literature.2
We report a coronavirus disease 2019
(COVID-19)–positive patient who presented with
severe abdominal pain and back pain and who was
subsequently diagnosed with a mycotic aortoiliac
aneurysm. Repeated blood and stool cultures
yielded S typhi. He was successfully treated with
endovascular stent graft repair and by postoperative
long-term antibiotics.
Case presentation
A 56-year-old frail malnourished man with a history
of diabetes mellitus presented with a 3-week history
of progressively worsening malaise and abdominal
and back pain. He had a fever of 38.1°C. Clinical
examination revealed tenderness over the central
and left lower quadrant of the abdomen. Initial
blood tests noted a haemoglobin level of 13.5 g/dL
and an elevated total white cell count at 13.58 × 109.
Chest radiograph was unremarkable, but emergency
computed tomography scan demonstrated a
saccular aortoiliac aneurysm involving the distal
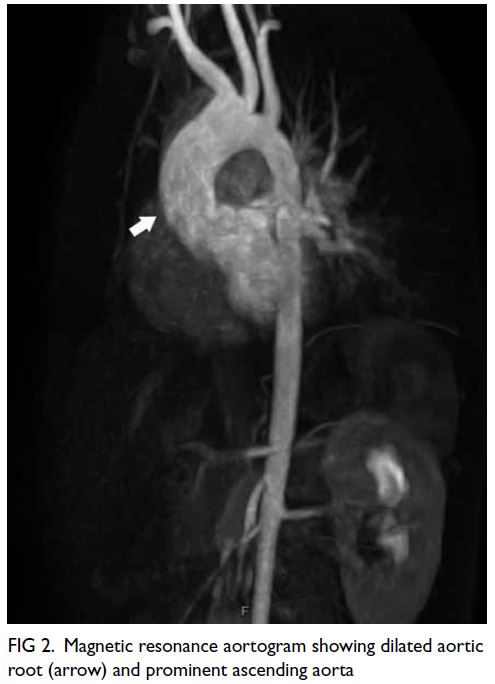
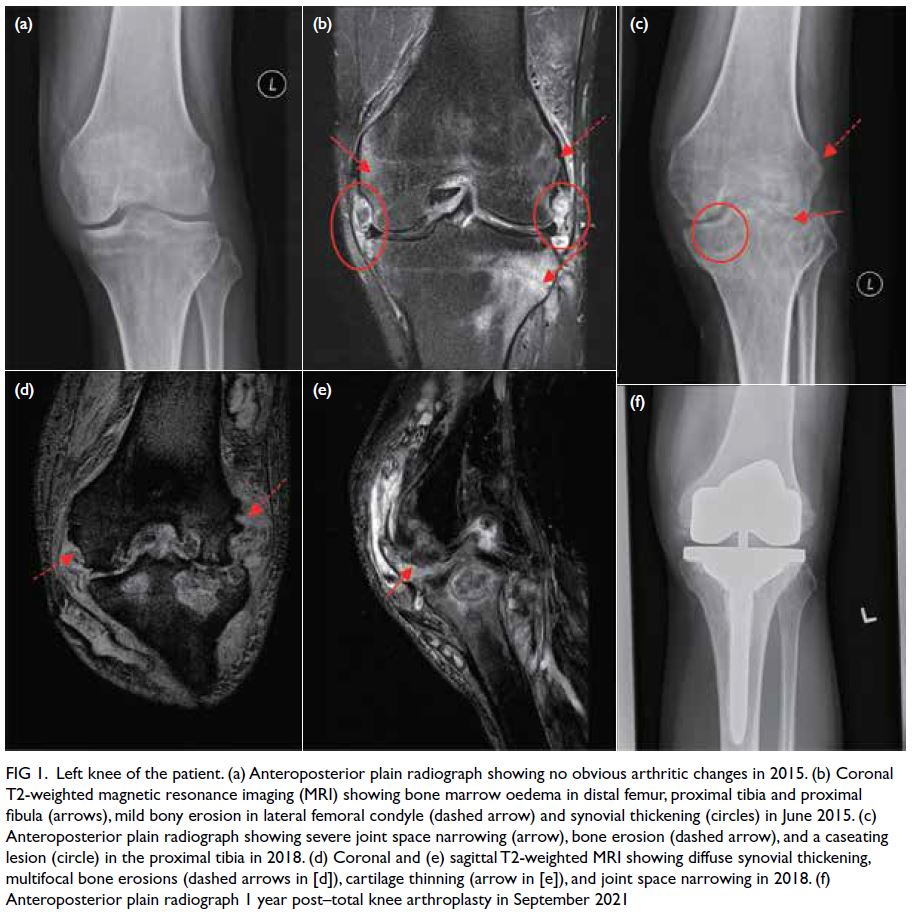
aorta and the left common iliac artery (Fig a and b).
Blood cultures were repeatedly positive for S typhi.
The patient was also tested positive for severe
acute respiratory syndrome coronavirus 2 (SARS-CoV-2) on admission screening necessitating patient
isolation.
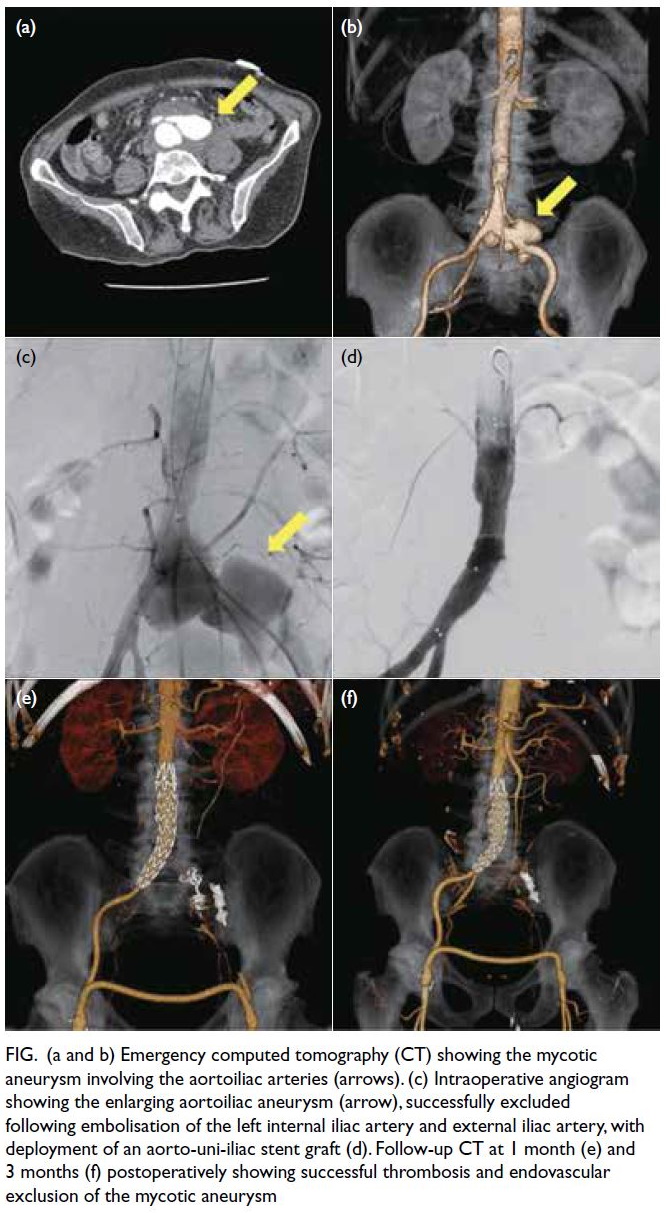
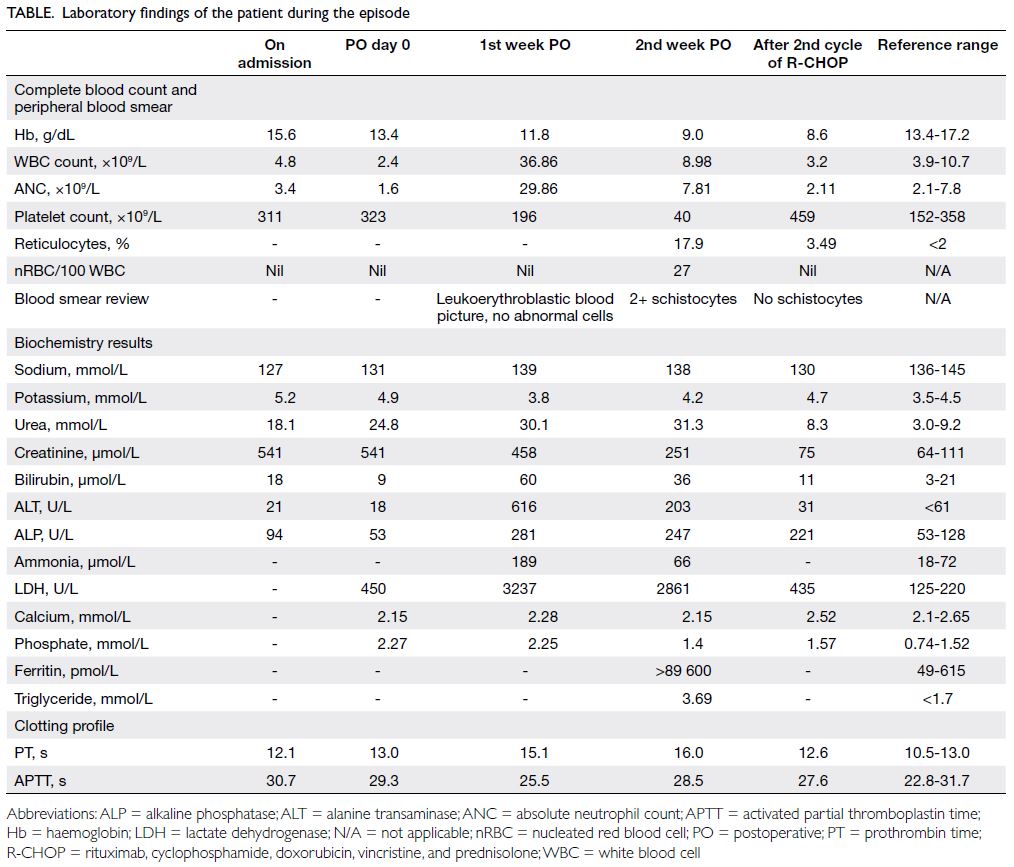
Figure. (a and b) Emergency computed tomography (CT) showing the mycotic aneurysm involving the aortoiliac arteries (arrows). (c) Intraoperative angiogram showing the enlarging aortoiliac aneurysm (arrow), successfully excluded following embolisation of the left internal iliac artery and external iliac artery, with deployment of an aorto-uni-iliac stent graft (d). Follow-up CT at 1 month (e) and 3 months (f) postoperatively showing successful thrombosis and endovascular exclusion of the mycotic aneurysm
Infection control specialists and microbiologists
were consulted for multidisciplinary management.
The patient was started on intravenous meropenem,
and fever resolved after 24 hours. Nonetheless in
view of persistent symptoms, emergent endovascular
intervention was performed 3 days following
admission. Angiogram confirmed the position and extent of the mycotic aneurysm (Fig c). The aortic
bifurcation was narrow (14.6 mm × 10.8 mm) and
the aortic diameter was small at 16 mm. Thus, we
opted to use an aorto-uni-iliac stent graft (Endurant
II; Medtronic, Galway, Ireland). The distal end
of the stent graft was deployed at the distal right
common iliac artery. The left internal iliac artery
was embolised with a 6 mm × 20 mm coil (Interlock
detachable coils; Boston Scientific, Marlborough
[MA], US), and the left external iliac artery with
an Amplatzer vascular plug (Abbott, Abbott Park
[IL], US). The left lower limb was revascularised
with a femoral-femoral bypass using a 7-mm ringed
polytetrafluoroethylene vascular graft (Advanta
VXT; Getinge, Gothenburg, Sweden). Completion
angiogram showed successful exclusion of the aortic
and left common iliac artery aneurysm (Fig d). The
patient’s discomfort improved. He was prescribed
intravenous meropenem for 6 weeks followed by
lifelong oral azithromycin based on culture sensitivity
results and the recommendation of microbiologists.
Follow-up computed tomography scan at 1 month
(Fig e) and 3 months (Fig f) postoperatively showed
thrombosis and successful endovascular exclusion of
the mycotic aneurysm.
Discussion
To the best of our knowledge this is the first
published case of a COVID-19–positive patient with
S typhi aortoiliac mycotic aneurysm. Salmonella
typhi is responsible for typhoid fever that is still
endemic in some South Asian and African countries.
Symptoms are primarily gastrointestinal with nausea
and diarrhoea but extraintestinal complications such
as aortitis and endocarditis may occur in the elderly
or immunocompromised individuals.3
Salmonella typhi is an exceedingly rare cause
of mycotic aneurysm. Only one previous report
has suggested the bacteria as a culprit. Guo et al2
showed that most Salmonella mycotic aneurysms
were caused by non-typhoidal Salmonella species
such as Salmonella enteritidis (30%) and Salmonella
choleraesuis (20%) with S typhi responsible for only
2% of cases in this cohort. The exact mechanism of typhoid-related aortic infection is unknown, but
possibilities include bacteraemia following bacterial
invasion of the gut mucosa, with seeding to the aortic
wall and subsequent aneurysmal degeneration.
Gallstones may provide a nidus for persistent
infection with possible contiguous spread to nearby
vasculature. The persistence of S typhi in mesenteric lymph nodes may contribute to relapsing typhoid
fever, resulting in disseminated infection long after
the initial presentation.4 Presumably, the presence of
S typhi in the para-aortic and para-iliac lymph nodes
could erode to the surrounding blood vessels with
subsequent aneurysm development.
Our patient was noted to be positive for
SARS-CoV-2 (ie, COVID-19 positive) during routine
admission screening. This may have important
implications in the pathogenesis, diagnosis and
subsequent management of many diseases. Due
to the ubiquitous spread of the virus, there have
been increasing reports of concurrent infection of
SARS-CoV-2 with local endemic pathogens. A
case report in 20215 documented co-infection with
both SARS-CoV-2 and S typhi in a 14-year-old boy
returning to Canada from Pakistan. This case report
concluded that since the potential presentation
of both disease entities would include fever and
gastrointestinal disturbance, the presence of
COVID-19 may have confounded the diagnosis
of other infections for clinicians unfamiliar with
typhoid fever. This diagnostic bias has also been
observed in countries where typhoid fever is
endemic with cases of early typhoid fever initially
treated as COVID-19 infection. This resulted in a
delay in prescribing appropriate antibiotic treatment
and possible progression to life-threatening
complications such as intestinal perforation.6 The
co-epidemic of COVID-19 and S typhi in some Asian
countries placed a heavy burden on local health care
resources. Insufficient medical resources combined
with delayed diagnosis and treatment contributed to
increased mortality from typhoid fever in patients
who may previously have made an uneventful
recovery.
Co-infection with SARS-CoV-2 and S typhi
may contribute to the pathogenesis and progression
of mycotic aneurysm. The relationship between
SARS-CoV-2 and mycotic aneurysms may be
explained by the immunosuppressive as well as
pro-inflammatory effects of COVID-19 infection.
Tian et al7 demonstrated that SARS-CoV-2 caused
immunosuppression in the early stages of infection
via suppression of chemokine signalling and immune
cell response. In our patient, immunosuppression
due to diabetes mellitus and recent COVID-19
infection may have resulted in increased risk of
bacterial seeding and colonisation of the aorta.
Contemporary literature also suggests that
COVID-19 may contribute to aneurysmal
development by upregulation and elaboration
of proinflammatory mediators and chemokines
by binding to angiotensin-converting enzyme 2
receptors on host cells.8 Current concepts of SARS-CoV-2 infection and aneurysm pathogenicity
suggest that COVID-19 may theoretically augment progression of aneurysms, the impact of which may
become clearer as the pandemic progresses.
Open operative management of mycotic
aneurysm has been gradually replaced by
endovascular options, with many studies
demonstrating effectiveness and durability of the
latter.9 Riley and Teixeira10 documented long-term
durability of endovascular intervention in
infected pseudoaneurysms. In our patient, we were
able to successfully exclude the aneurysm with an
endovascular approach, but long-term follow-up
and surveillance imaging is essential to guarantee
durability. In terms of his COVID-19 infection, the
patient did not develop any respiratory symptoms nor
were any abnormalities noted on chest radiographs,
and no specific therapy was provided.
In conclusion, typhoid fever remains a major
worldwide public health concern. This is the first
case in the world’s contemporary literature of a
S typhi mycotic aortoiliac aneurysm in a patient
infected with COVID-19. This report emphasises
the importance of early tertiary vascular referral and
prompt multidisciplinary management involving
microbiologists, infection control specialists, and
vascular surgeons. We were able to control the
sepsis with timely endovascular treatment yielding
good mid-term results. Although the global
situation regarding COVID-19 is improving and
our understanding of COVID-19 is increasingly well
developed, this case report aims to raise awareness
among readers about the possible impact of
COVID-19 on the diagnosis, presentation, and
development of other pathologies.
Author contributions
Concept or design: YC Chan.
Acquisition of data: SE So.
Analysis or interpretation of data: SE So.
Drafting of the manuscript: SE So.
Critical revision of the manuscript for important intellectual content: YC Chan, SW Cheng.
Acquisition of data: SE So.
Analysis or interpretation of data: SE So.
Drafting of the manuscript: SE So.
Critical revision of the manuscript for important intellectual content: YC Chan, SW Cheng.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have declared no conflicts of interests.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of Helsinki and provided informed written consent to publication of this report.
References
1. Taylor PR, Chan YC. Endovascular treatment in the
management of mycotic aortic aneurysms. In: Thompson
MM, Morgan RA, Matsumara JS, Sapoval M, Loftus I,
editors. Endovascular Intervention for Vascular Disease.
Principles and Practice. London: Informa Healthcare;
2008: 235-41.
2. Guo Y, Bai Y, Yang C, Wang P, Gu L. Mycotic aneurysm due
to Salmonella species: clinical experiences and review of
the literature. Braz J Med Biol Res 2018;51:e6864. Crossref
3. Griffin AJ, Li LX, Voedisch S, Pabst O, McSorley SJ.
Dissemination of persistent intestinal bacteria via the
mesenteric lymph nodes causes typhoid relapse. Infect
Immun 2011;79:1479-88. Crossref
4. Hohmann EL. Nontyphoidal salmonellosis. Clin Infect Dis
2001;32:263-9. Crossref
5. Ayoubzadeh SI, Isabel S, Coomes EA, Morris SK. Enteric
fever and COVID-19 co-infection in a teenager returning
from Pakistan. J Travel Med 2021;28:taab019. Crossref
6. Abdul Aziz JM, Abdullah SK, Al-Ahdal TM, et al.
Diagnostic bias during the COVID-19. A rare case report
of Salmonella typhi. Ann Med Surg (Lond) 2022:74:103282. Crossref
7. Tian W, Zhang N, Jin R, et al. Immune suppression in
the early stage of COVID-19 disease. Nat Commun
2020;11:5859. Crossref
8. Xu B, Li G, Guo J, et al. Angiotensin-converting enzyme 2,
coronavirus disease 2019, and abdominal aortic aneurysms.
J Vasc Surg 2021;74:1740-51. Crossref
9. Sörelius K, Mani K, Björck M, et al. Endovascular treatment
of mycotic aortic aneurysms: a European multicenter
study. Circulation 2014;130:2136-42. Crossref
10. Riley CJ, Teixeira P. Development of symptomatic
inflammatory aneurysm treated with endovascular repair
in coronavirus disease 2019–infected patient. J Vasc Surg
Cases Innov Tech 2021;7:193-6. Crossref