Progressively enlarged Caesarean section scar endometriosis during pregnancy: case report and literature review
Hong Kong Med J 2026;32:Epub 28 Apr 2026
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Progressively enlarged Caesarean section scar
endometriosis during pregnancy: case report and literature review
Yushi Wu, MD1,2; Xiaoyan Li, MD1,2; Jinghua Shi, MD1,2; Yuyuan Li1,2; Junhua Leng, MD1,2; Yi Dai, MD1,2
1 Department of Obstetrics and Gynecology, Peking Union Medical College Hospital, Chinese Academy of Medical Science and Peking Union Medical College, Beijing, China
2 National Clinical Research Center for Obstetrics and Gynecologic Diseases, Beijing, China
Corresponding author: Prof Yi Dai (jacquedai2024@163.com)
 Full paper in PDF
Full paper in PDF
Case presentation
A 36-year-old full-term pregnant woman with a
history of prior Caesarean section (CS) presented
to Peking Union Medical College Hospital in
November 2024 for a planned repeat CS. In 2017, she
underwent an emergency CS due to arrest of labour.
One year after surgery, ultrasound revealed a 1-cm
nodule near the incision, suspected to be abdominal
wall endometriosis (AWE). The patient declined
intervention at that time. On subsequent follow-up,
the mass exhibited progressive enlargement and
multifocal spread. Since 2019, she had experienced tenderness and aggravated menstrual-associated
pain. Preconception evaluation documented a body
mass index of 24.14 kg/m2 and a 5-cm mass. She had
conceived naturally. During pregnancy, the mass
demonstrated continued growth, with accelerated
expansion noted after 30 weeks of gestation. At 36
weeks of gestation, outpatient assessment revealed a
cystic mass measuring 10 × 6 cm2 in the subcutaneous
tissue of the left scar margin, with focal bluish
discolouration of the overlying skin. Ultrasound
revealed two confluent masses with a total size of
9.9 × 4.5 × 2.6 cm3, with prominent internal
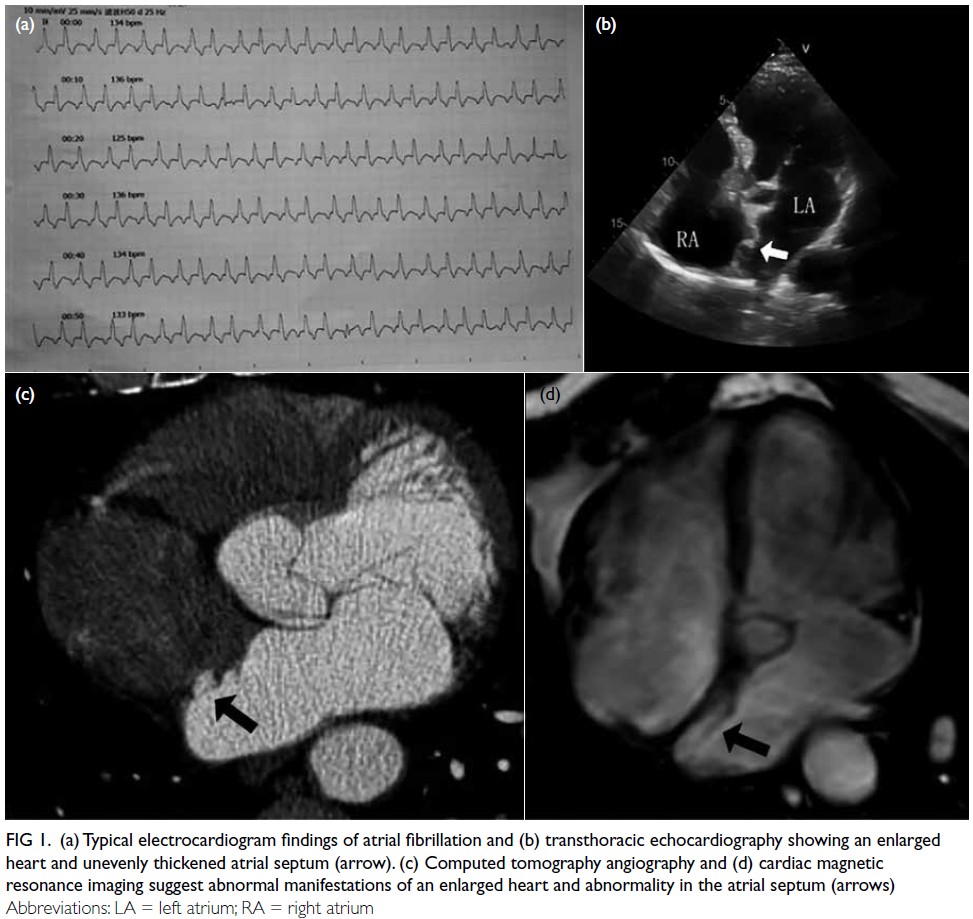
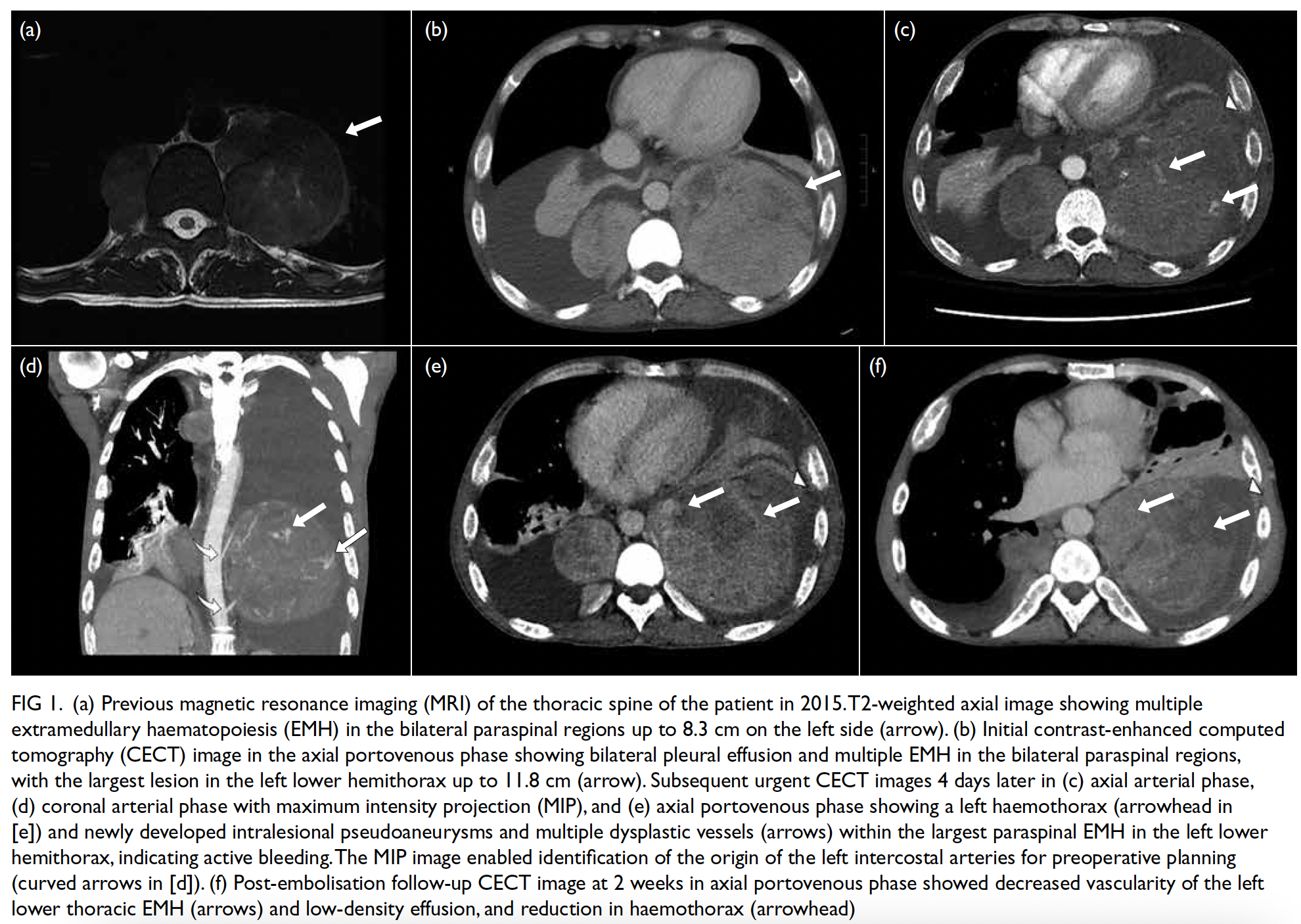
vascularity (Fig 1).
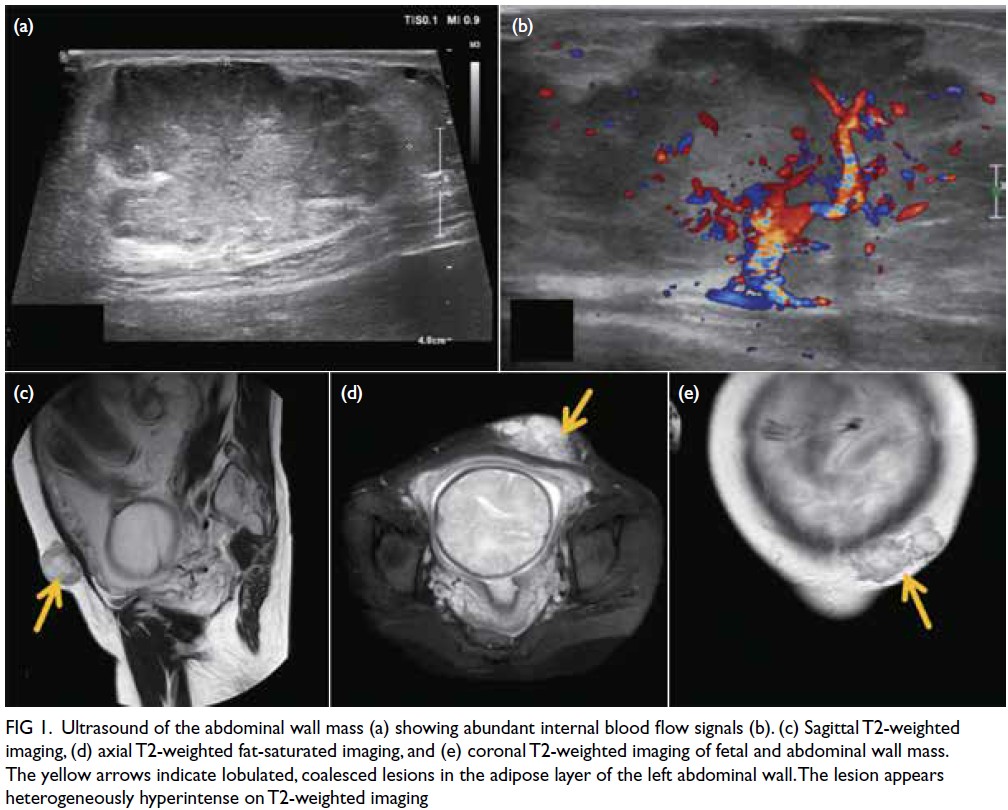
Figure 1. Ultrasound of the abdominal wall mass (a) showing abundant internal blood flow signals (b). (c) Sagittal T2-weighted imaging, (d) axial T2-weighted fat-saturated imaging, and (e) coronal T2-weighted imaging of fetal and abdominal wall mass. The yellow arrows indicate lobulated, coalesced lesions in the adipose layer of the left abdominal wall. The lesion appears heterogeneously hyperintense on T2-weighted imaging
Preoperative magnetic resonance imaging
(MRI) was performed to evaluate the depth of
invasion, revealing two lobulated subcutaneous
masses exhibiting mixed long T1 and slightly long
T2 signals, with an invasion depth of 3.6 cm (Fig 1).
The patient underwent elective CS under spinal
anaesthesia. The original surgical scar was excised
to ensure adequate exposure of the mass, which
measured over 10 cm in diameter and extended
from the subcutaneous tissue to the anterior sheath
of the rectus abdominis. The lesion was carefully
dissected along its superior margin under manual
guidance. Once sufficient space was created,
Caesarean delivery was performed. The patient was
converted to general anaesthesia after delivery and
the abdominal wall lesion was completely excised.
An en bloc resection of the lesion was performed to
avoid fragmentation and potential dissemination. In
addition, the abdominal wall wound was thoroughly
irrigated to minimise the risk of implantation of
endometriotic tissue. Intraoperative exploration of
the pelvic cavity showed no evidence of concurrent
pelvic endometriosis. The 40-minute resection was
followed by primary abdominal wall reconstruction
without mesh. Subcutaneous drainage and
compression dressings were applied. No antibiotics
were administered and the patient remained afebrile. The drain was removed 3 days after surgery and the
wound achieved primary healing. Both the patient
and the neonate had no complications and were
discharged 5 days postoperatively.
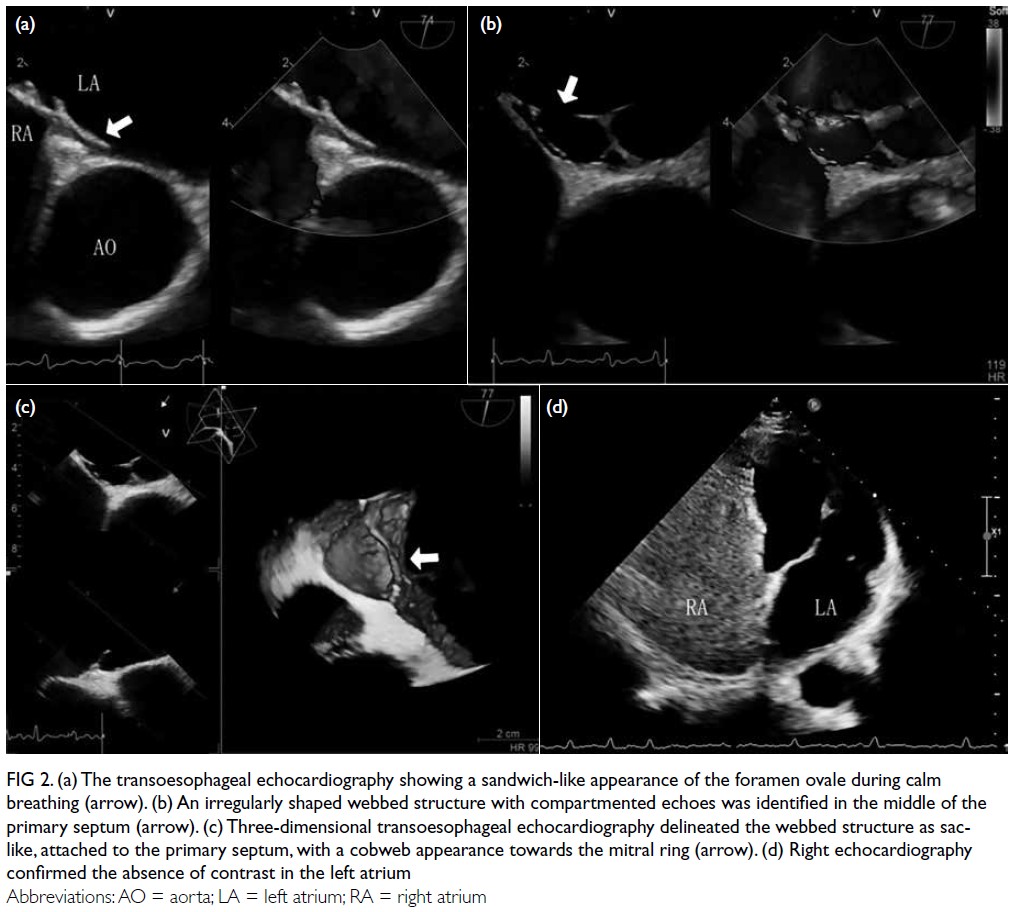
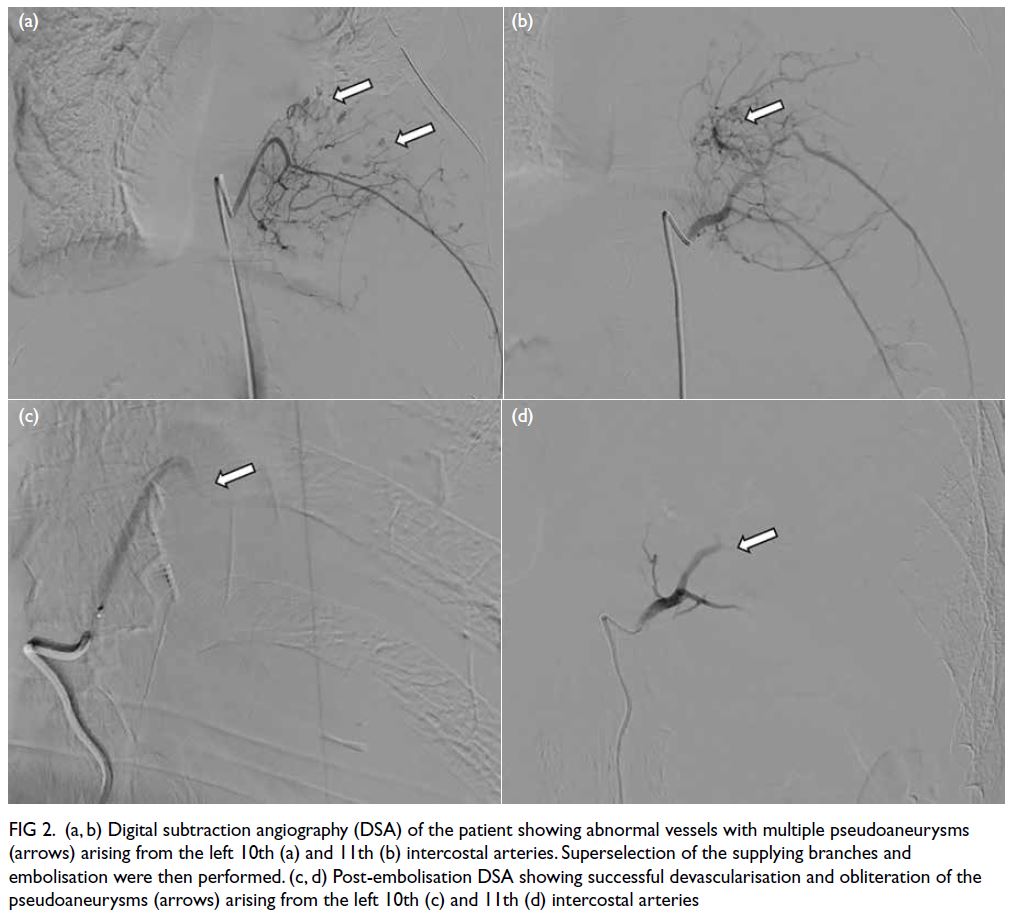
Histopathological examination confirmed
that the lesion was consistent with endometriosis,
showing decidual-like changes in the stroma and
glandular atrophy with a tubular appearance.
Immunohistochemistry revealed the lesion to
be oestrogen receptor–negative and stromal
progesterone receptor–positive (Fig 2).
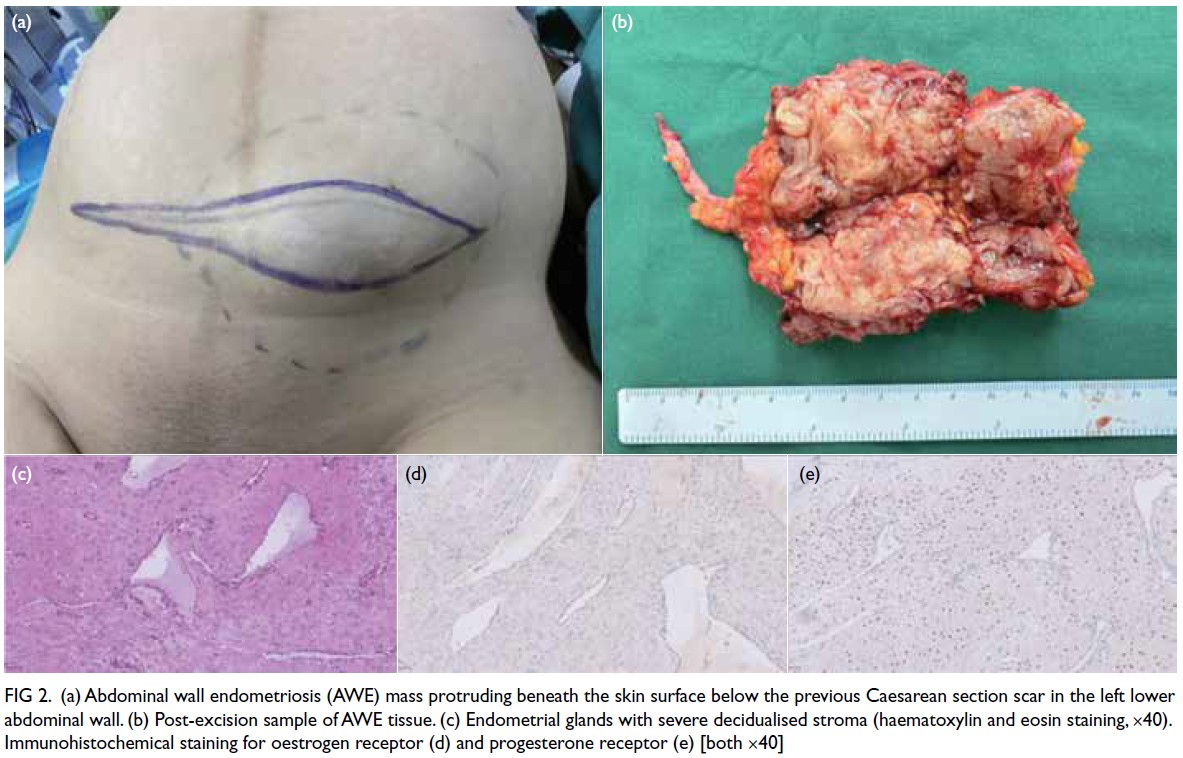
Figure 2. (a) Abdominal wall endometriosis (AWE) mass protruding beneath the skin surface below the previous Caesarean section scar in the left lower abdominal wall. (b) Post-excision sample of AWE tissue. (c) Endometrial glands with severe decidualised stroma (haematoxylin and eosin staining, ×40). Immunohistochemical staining for oestrogen receptor (d) and progesterone receptor (e) [both ×40]
Discussion
Abdominal wall endometriosis is a rare form
of endometriosis, with a reported incidence of
0.03% to 0.4%.1 Most cases occur near a CS scar.
Previous studies have described changes in pelvic
endometriosis during pregnancy,2 3 but there is
limited literature describing the progression and
management of AWE during pregnancy. In this case,
the patient experienced significant enlargement of
scar endometriosis during pregnancy, indicating
that AWE, similar to pelvic endometriotic lesions,
may also gradually enlarge during gestation. The
first-line treatment for AWE is surgical excision of
the mass, which is typically performed in the non-pregnant period. As the lesion is often closely related
to a previous CS scar, simultaneous excision may
significantly increase the complexity of CS. In this
case, the lesion was dissected along its margin during
CS. Our retrospective study demonstrated that
among 367 patients who underwent margin-based
excision, the recurrence rate was 3.3%,4 comparable
to or lower than rates reported in previous studies.5 6
This suggests that excision along the lesion margin
is sufficient and does not increase the risk of
recurrence. After thorough assessment, excision
during CS may be considered if the lesion is small and
demonstrates minimal depth of invasion. Given the
potential for lesion enlargement and the challenges
of abdominal wall reconstruction during pregnancy,
timely surgical intervention is particularly advisable
for patients planning future pregnancies.
Preoperative evaluation is also crucial. Our
previous research classified patients into three
types according to the depth of lesion invasion.4
This patient could be clinically classified as Type I
AWE. Given the minimal muscular invasion and
stretching of the abdominal wall due to the gravid
uterus, primary closure without mesh placement
was feasible. This highlights the importance of
comprehensive preoperative assessment to guide the
choice of surgical approach and anticipate operative
complexity.
Decidualisation of endometriosis can occur
under the influence of high progesterone levels.7
Since Pellegrini8 first described this condition in
1982, only nine cases of decidualised AWE have
been reported.2 8 9 10 11 12 13 14 15 In these cases, the mean age was
28.6 ± 5.8 years, and the mean latency from first CS
to mass presentation was 29.6 ± 24.2 months. The
depth of invasion in these patients differs from that
in non-pregnant patients. Our previous research4
showed that in non-pregnant patients, AWE lesions
most commonly invade the rectus abdominis
muscle. In contrast, existing literature indicates
that in pregnant patients, lesions were confined
to the subcutaneous and adipose layers, with only
one case showing invasion of the rectus abdominis.
Enlargement during pregnancy was reported in
two patients, with the maximum diameter reaching
3 cm.9 10 Six patients underwent lesion excision
during CS,2 8 10 11 12 13 and one underwent elective surgery
after CS.14 In our case, the patient experienced
rapid enlargement of the AWE during pregnancy
and imaging raised a suspicion of malignancy.
This suggests that decidualised AWE during
pregnancy may present with features mimicking
malignancy. Histopathological examination in our
patient revealed a positive progesterone receptor
expression and absence of oestrogen receptor
expression, indicating that the intrinsic biological
characteristics of the lesion may have contributed
to its abnormal growth. Limited treatment options during pregnancy pose additional risks to maternal
and fetal safety. Therefore, in patients with AWE
who wish to conceive in the future, surgery should
ideally be performed before pregnancy.
Author contributions
Concept or design: Y Wu, J Leng, Y Dai.
Acquisition of data: Y Wu, J Shi, X Li..
Analysis or interpretation of data: Y Li..
Drafting of the manuscript: Y Wu..
Critical revision of the manuscript for important intellectual content: J Leng, Y Dai.
Acquisition of data: Y Wu, J Shi, X Li..
Analysis or interpretation of data: Y Li..
Drafting of the manuscript: Y Wu..
Critical revision of the manuscript for important intellectual content: J Leng, Y Dai.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study was supported by the National Key Research and
Development Program Project (Ref No.: 2022YFC2704000)
and the National Natural Science Foundation of China (Ref
No.82071628). The funders had no role in the study design,
data collection/analysis/interpretation, or manuscript
preparation.
Ethics approval
This study was approved by the Institutional Review Board
of Peking Union Medical College Hospital, China (Ref No.:
K23N4099). Informed consent was obtained from the patient
for all treatments and procedures, and for publication of the
case report with the accompanying clinical images.
References
1. Carsote M, Terzea DC, Valea A, Gheorghisan-Galateanu AA. Abdominal wall endometriosis (a narrative review). Int J Med Sci 2020;17:536-42.
Crossref
2. D’Agostino C, Surico D, Monga G, Palicelli A. Pregnancy-related decidualization of subcutaneous endometriosis occurring in a post–Caesarean section scar: case study and review of the literature. Pathol Res Pract 2019;215:828-31.
Crossref
3. Sachdeva G, Divyashree PS, Shailaja N. A non-classical presentation of scar endometriosis during pregnancy: case report and review of literature. JBRA Assist Reprod 2022;26:563-66.
Crossref
4. Wu Y, Dai Y, Zhang J, et al. The clinical features and long-term surgical outcomes of different types of abdominal wall endometriosis. Arch Gynecol Obstet 2023;307:163-8.
Crossref
5. Marras S, Pluchino N, Petignat P, et al. Abdominal wall endometriosis: an 11-year retrospective observational cohort study. Eur J Obstet Gynecol Reprod Biol X 2019;4:100096.
Crossref
6. Horton JD, Dezee KJ, Ahnfeldt EP, Wagner M. Abdominal wall endometriosis: a surgeon’s perspective and review of 445 cases. Am J Surg 2008;196:207-12.
Crossref
7. Frühauf F, Fanta M, Burgetová A, Fischerová D. Endometriosis in pregnancy—diagnostics and management. Ceska Gynekol 2019;84:61-7.
8. Pellegrini AE. Cutaneous decidualized endometriosis. A pseudomalignancy. Am J Dermatopathol 1982;4:171-4.
Crossref
9. El-Gohary YM, Garcia MT, Ganjei-Azar P. Decidualized endometrioma diagnosed by fine needle aspiration cytology: a case report with immunocytochemical confirmation. Diagn Cytopathol 2009;37:373-6.
Crossref
10. Nogales FF, Martin F, Linares J, Naranjo R, Concha A. Myxoid change in decidualized scar endometriosis mimicking malignancy. J Cutan Pathol 1993;20:87-91.
Crossref
11. Kavari A, Samizadeh B, Maghbool M. Pregnancy-related decidualization in post–Cesarean section scar endometriosis: a case report. Int J Surg Case Rep 2024;124:110274.
Crossref
12. Natale KE, Royer MC, Rush WL, Lupton GP. Cutaneous deciduosis: a report of two cases of an unusual pseudomalignancy. J Cutan Pathol 2012;39:777-80.
Crossref
13. Günal A, Keskin U, Deveci G, Deveci MS, Yenen MC, Dede M. Tumor-like myxoid change in decidualized scar endometriosis of pregnancy: a case report and review of literature. Turk J Pathol 2009;25:49-52.
Crossref
14. Val-Bernal JF, Val D, Gómez-Aguado F, Corcuera MT, Garijo MF. Hypodermal decidualized endometrioma with aberrant cytokeratin expression. A lesion mimicking malignancy. Am J Dermatopathol 2011;33:e58-62.
Crossref
15. Berardo M, Valente P, Powers C. Cytodiagnosis and comparison of nondecidualized and decidualized endometriosis of the abdominal wall. A report of two cases. Acta Cytol 1992;36:957-62.
 Three video clips showing the webbed left atrial septal pouch and contrast flow are available at
Three video clips showing the webbed left atrial septal pouch and contrast flow are available at