Xanthogranulomatous inflammation of terminal ileum: report of a case with small bowel involvement
DOI: 10.12809/hkmj134103
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Xanthogranulomatous inflammation of terminal ileum: report of a case with small bowel
involvement
KC Wong, MCSHK, MRCSEd1;
Wilson MS Tsui, FIAC, FRCPath2;
SJ Chang, FRCS, FRCSEd1
1 Department of Surgery, Caritas Medical Centre, Shamshuipo, Hong Kong
2 Department of Pathology, Caritas Medical Centre, Shamshuipo, Hong Kong
Corresponding author: Dr KC Wong(kamkam44@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Xanthogranulomatous inflammation is a rare
pathological condition most frequently detected
in the kidney and gallbladder. Reported herein is
a case of xanthogranulomatous inflammation in
a 51-year-old male presenting as a mass-forming
lesion in the terminal ileum with mucosal ulceration.
Diagnostic laparoscopy followed by ileocecectomy
was performed due to intra-operative suspicion
of carcinoma of appendix. This is a report of
the condition involving the terminal ileum with
mucosal ulceration and full-thickness involvement
of bowel wall which are uncommon features of
xanthogranulomatous inflammation in previously
reported lower gastro-intestinal tract lesions.
Introduction
Xanthogranulomatous inflammation (XGI) is a
rare but well-defined disease, first reported by
Oberling in 1935.1 The disease process was most
frequently reported in the kidney and gallbladder.
Rare occurrence in the gastro-intestinal tract was
illustrated in only one recently reported case in the
terminal ileum,2 four reported cases in the colon,3 4 5 6 eight cases in a series of interval appendicectomy
specimens,7 and eight cases with gastric
involvement.8 9 10 11 12 13 Of the four cases with colonic
involvement, two involved the sigmoid colon,3 4 one involved the caecum,5 and one involved the ascending
colon.6 Most of these colonic lesions presented with a
mass-forming lesion with predominant submucosal
involvement, while primary mucosal involvement
was only reported in the last case involving the
ascending colon. We, herein, report the second case
of XGI in the terminal ileum with mucosal ulceration
and full-thickness involvement of the bowel wall,
presenting as a painful right-lower-quadrant
abdominal mass.
Case report
A 51-year-old Chinese male presented to the
Emergency Department on 2 December 2012. He
was a chronic smoker and alcoholic. He complained
of right-sided abdominal pain for the past 2 weeks.
The pain was not associated with nausea, vomiting,
constipation, or diarrhoea. There was no anorexia
or weight loss. There was no history of melaena. His
medical history included diabetes, hypertension,
and gout. There was no history of tuberculosis.
He was admitted to a hospital in Mainland
China 10 days before the index admission for the
same problem. While in that hospital, he had raised
white cell count of 16.2 x 109 /L, and an ultrasound
of abdomen revealed a gallstone and a renal stone.
There was no hydronephrosis. A course of antibiotics
was given, but the symptoms persisted. The patient
returned from Mainland China on 2 December
2012, and attended our Emergency Department for
further management.
On admission, the physical examination of
the respiratory, cardiovascular, and central nervous
systems was unremarkable. Abdominal examination
revealed tenderness in the right lower quadrant.
Per rectal examination revealed no blood, melaena,
or mass. Abdominal X-ray showed no specific
abnormalities.
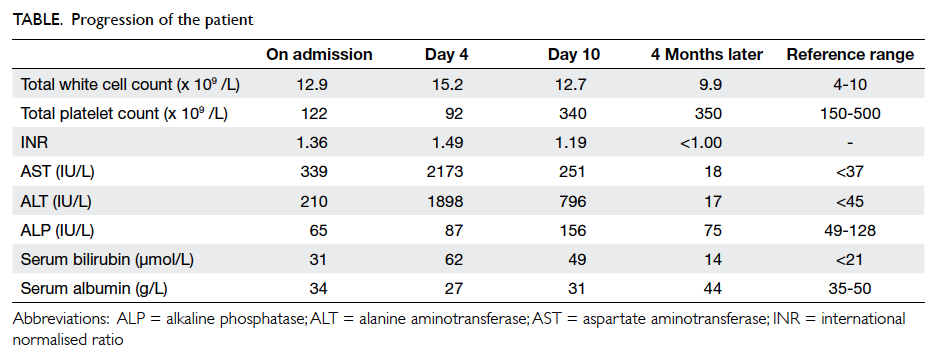
His blood tests revealed mildly elevated
white cell count of 10.6 x 109 /L (reference range
[RR], 3.7-9.2 x 109 /L), haemoglobin level of 137 g/L
(RR, 134-171 g/L), normal amylase level of 57 U/L
(RR, 30-128 U/L), and normal electrolytes and liver
enzymes. An urgent ultrasound of the abdomen
and pelvis raised suspicion of acute appendicitis,
with a tubular, non-peristaltic, non-compressible
structure measuring 1.55 cm in diameter at the
appendicular region, with small amount of loculated
fluid around the lesion (Fig 1), corresponding with
the site of maximum tenderness. The distal end of
the lesion was obscured by bowel gas; it could not be
ascertained whether it was blind-ended. The same
study also revealed the presence of a gallstone and a
right lower pole renal stone.
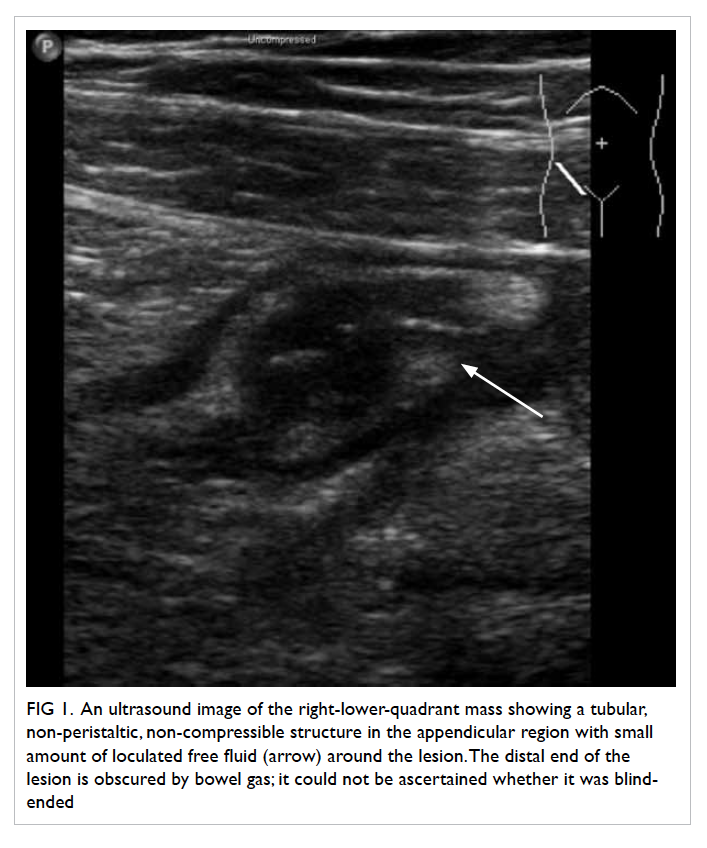
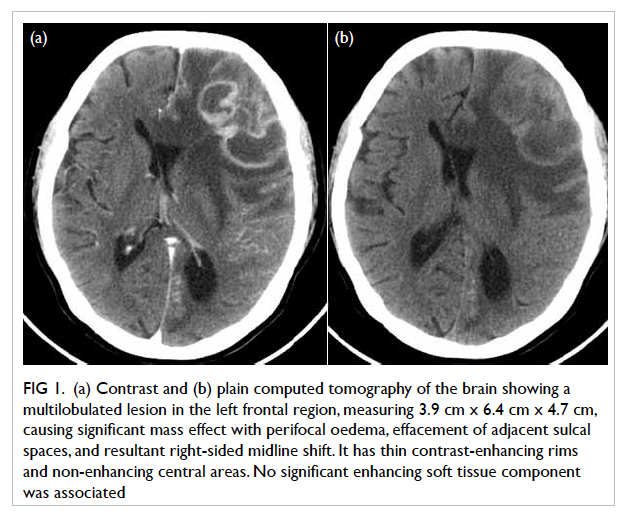
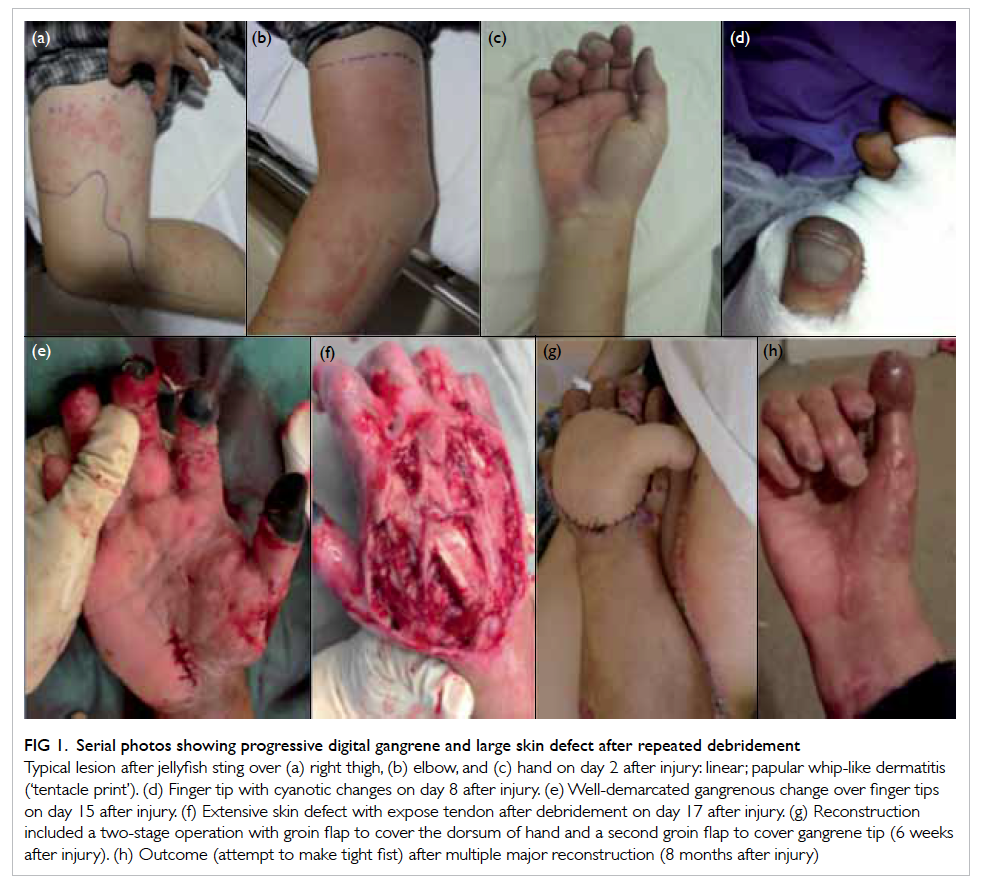
Figure 1. An ultrasound image of the right-lower-quadrant mass showing a tubular, non-peristaltic, non-compressible structure in the appendicular region with small amount of loculated free fluid (arrow) around the lesion. The distal end of the lesion is obscured by bowel gas; it could not be ascertained whether it was blind-ended
With a preliminary diagnosis of acute
appendicitis, diagnostic laparoscopy was performed
on 3 December 2012. There was an ileocaecal
mass fixed to the posterior abdominal wall, which
was difficult to mobilise despite an open approach
via gridiron incision. Upon conversion to midline
laparotomy, a large inflammatory mass was found
at the ileocaecal junction, compatible with an
infiltrative tumour. The mass demonstrated through-and-through invasion into the ileal mesentery,
involving several loops of the ileum. Ileocaecal
lymph node enlargement was noted. The appendix
was not identified. There was no gross cavity or
pus. With the suspicion of carcinoma of appendix,
limited right hemicolectomy with en-bloc resection
of the mass together with 65 cm of the terminal
ileum, caecum, and proximal ascending colon was
performed. Primary sub-end to sub-end, side-to-side
anastomosis was fashioned.
The postoperative course of the patient was
complicated by on-and-off fever with elevated white
cell count of up to 22.5 x 109 /L. Blood culture taken
on postoperative day 2 yielded no bacterial growth.
Erythema and serous discharge were noted in the
paraumbilical region of the laparotomy and gridiron
wounds, which were managed with dressing and
packing. Wound swab yielded scanty growth of
Escherichia coli. The fever responded to a 10-day
course of cefuroxime and metronidazole. He was
discharged on postoperative day 10.
A colonoscopy done 4 months after the
operation in April 2013 revealed no abnormalities.
Pathological examination
Gross examination of the ileocolectomy specimen
revealed a 0.5-cm ileal mucosal ulcer, which
was 1.5 cm proximal to the ileocaecal valve.
The periappendicular mass was haemorrhagic
and covered with exudate, within which was a
retrocaecal appendix, measuring 5 cm in length
and 1 cm in diameter, surrounded by necrotic and
yellowish tissue. Cut surface of the appendix was
unremarkable. A few lymph nodes were found in the
ileocaecal fossa.
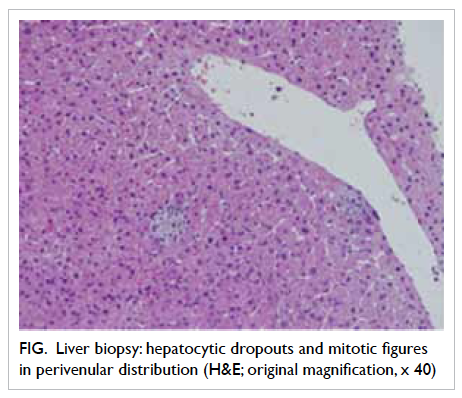
Microscopic examination of the appendicular
mass showed abscess, haemorrhage, and XGI which
consisted predominantly of foamy histiocytes,
scattered neutrophils, lymphoplasmacytic cells,
a few multinucleated giant cells, and congested
capillaries with surrounding fibrosis (Fig 2). The
foamy histiocytes were positive for CD68 on
immunostaining, confirming their histiocytic origin.
No Michaelis-Gutmann bodies were detected. The
terminal ileum ulcer revealed similar XGI which
extended through the bowel wall to involve the
mesentery. The appendicular mucosa showed no
neutrophilic infiltrate. A few reactive lymph nodes
were noted. There was no evidence of malignancy or
granuloma.
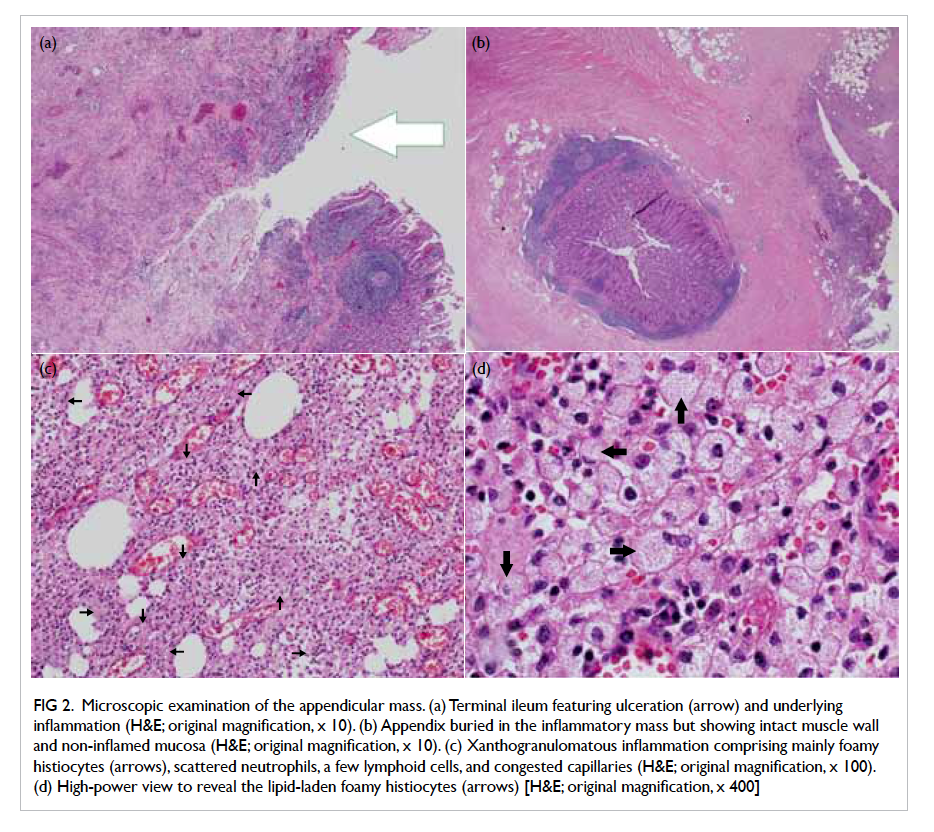
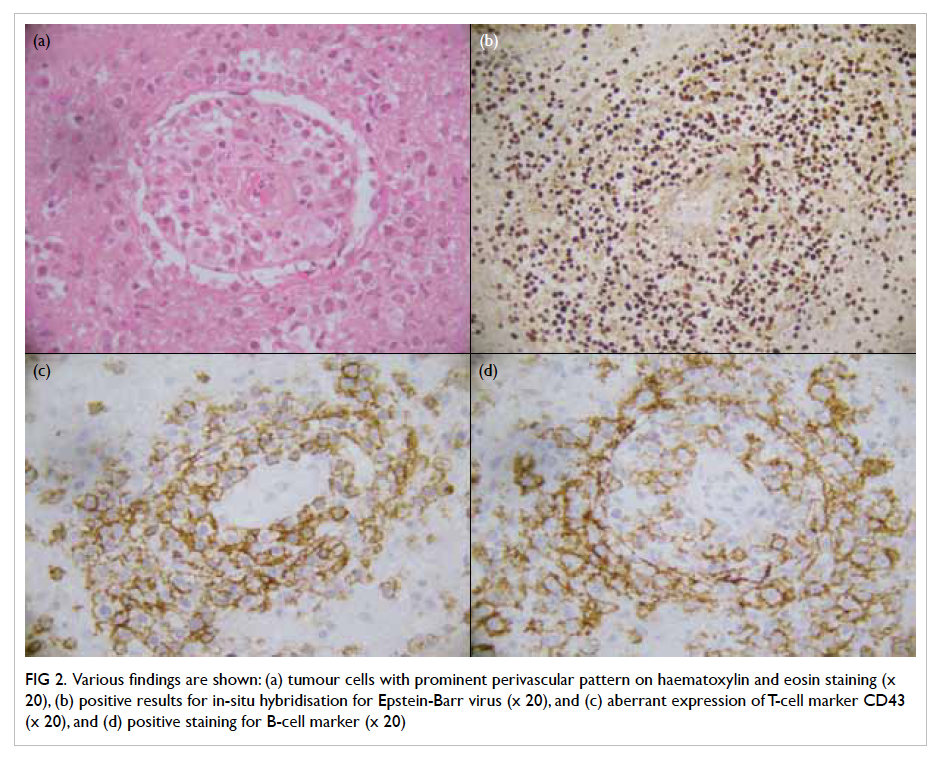
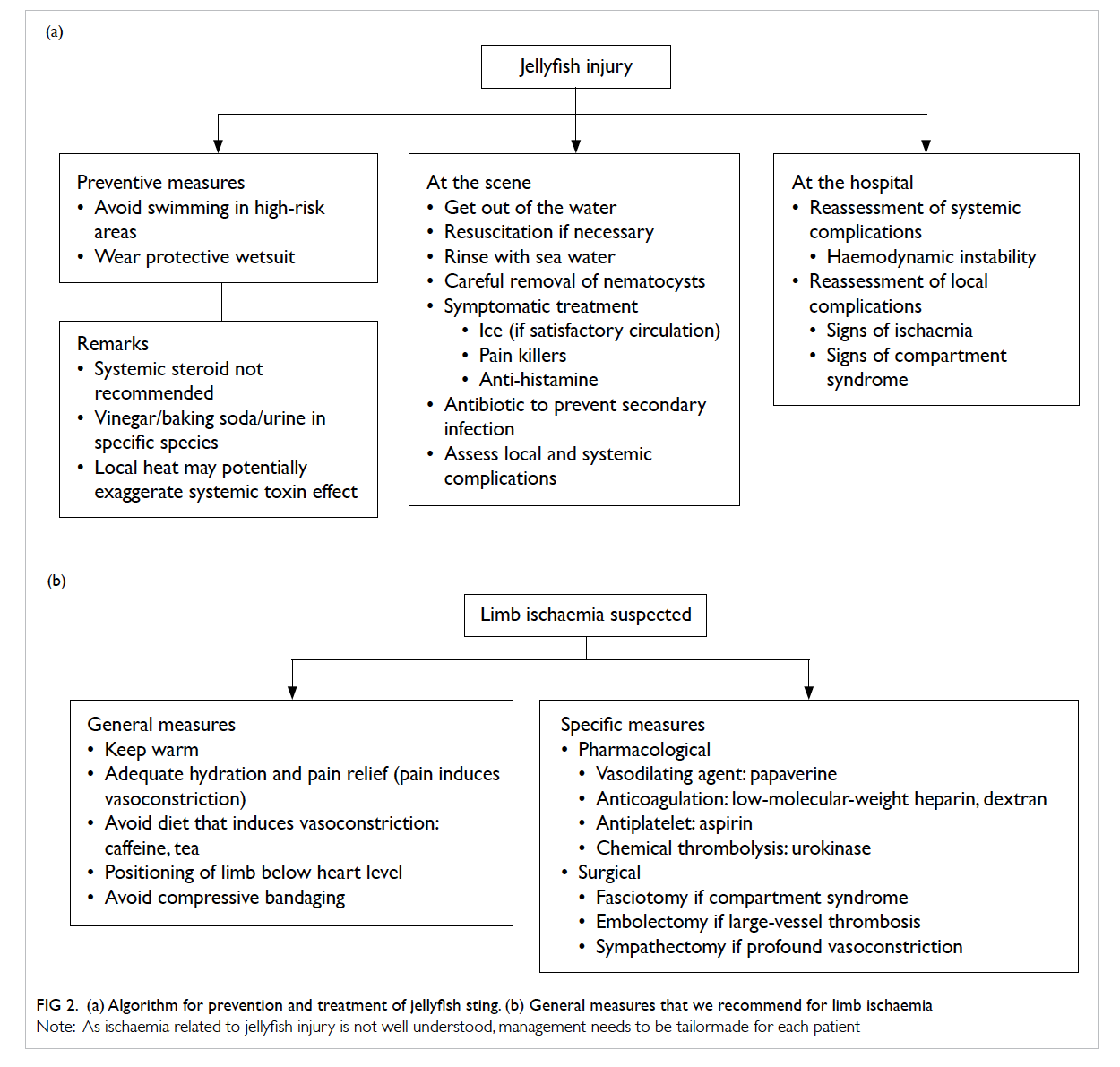
Figure 2. Microscopic examination of the appendicular mass. (a) Terminal ileum featuring ulceration (arrow) and underlying inflammation (H&E; original magnification, x 10). (b) Appendix buried in the inflammatory mass but showing intact muscle wall and non-inflamed mucosa (H&E; original magnification, x 10). (c) Xanthogranulomatous inflammation comprising mainly foamy histiocytes (arrows), scattered neutrophils, a few lymphoid cells, and congested capillaries (H&E; original magnification, x 100). (d) High-power view to reveal the lipid-laden foamy histiocytes (arrows) [H&E; original magnification, x 400]
Discussion
Xanthogranulomatous inflammation is a form of
chronic inflammatory condition characterised
macroscopically by mass-forming golden yellow
tumours and microscopically by aggregation of lipid-laden foamy histiocytes including multinucleated
giant cells, with a minor component of chronic and
acute inflammatory cells and fibrous reaction. It was
first described by Oberling in 1935 in three cases of
retroperitoneal xanthogranulomas.1 Its occurrence
in the endometrium, ovary, fallopian tubes, vagina,
testis, epididymis, stomach, bone, skin (as fistulation
secondary to inflammation primarily involving
another internal organ),14 appendix,7 15 urinary
bladder, thyroid, and adrenal glands has been
reported, with the highest prevalence reported in
the kidney and gallbladder. A majority of XGI cases
present as a mass-like lesion with an extension of
fibrosis and inflammation to the surrounding tissues,
leading to diagnostic difficulties in differentiating
them from infiltrative malignant tumours.
Pathological differential diagnoses bearing
similar histological features include malakoplakia,
which is characterised by an inflammatory and
destructive xanthomatous proliferation with the
presence of Michaelis-Gutmann bodies, which are
intracytoplasmic laminated concretions usually
positive for periodic acid–Schiff, von Kossa, and Prussian blue
stains. Macrophages known as von Hansemann
cells are more granular and eosinophilic and have
less vacuolated cytoplasm than ordinary histocytes.
Other differential diagnoses include localised
xanthoma deposits without parenchymal destruction
or xanthomas with prominent foam cell features.
Although the pathological features of XGI are
well described, its exact pathogenesis is not well
established.
Various proposed mechanisms include chronic
recurrent infection, obstruction, immunological
disorders, and defective lipid transport. It is generally
believed that the localised proliferation of lipid-laden
foamy histiocytes in XGI represents chronic
suppurative inflammation secondary to interaction
between the host and micro-organisms. Examples
of immunological disorders include disrupted
chemotaxis of polymorphs and macrophages, which
is a specific immune response toward Proteus and
Escherichia infections. A recently reported case2
involving the terminal ileum proposed a possible
mechanism of perforation due to an ingested foreign
body. However, none of the above hypotheses were
able to fully explain the anatomical distribution of the
condition, which is most common in the appendix
where neither perforation due to ingested foreign
bodies nor chronic suppurative inflammation is
most often found. In this reported case, infected
laparotomy wound swab yielded E coli, while there
were no symptoms to suggest pre-existing chronic
suppurative inflammation. There was no evidence
of a penetrating foreign body on history or gross
examination of the pathological specimen.
Rare occurrence of XGI in the lower gastro-intestinal
tract is illustrated by only one reported
case in the terminal ileum,2 four reported cases in
the colon with two involving the sigmoid colon,3 4 one involving the caecum,5 and one involving the
ascending colon.6 However, a histopathological
review of 22 interval appendicectomy specimens by Guo and Greenson7 in 2003 reported presence of XGI in eight cases (36.4%) of interval appendicectomy
versus none in the 44 matched patients receiving
acute appendicectomy, suggesting XGI may be
underreported as a delayed consequence of acute
inflammation and that these histological changes
are secondary to the time interval of inflammation
rather than intrinsic factors specific to the patient or
disease.
Due to endoscopic, radiological, and the intra-operative
macroscopic resemblance to infiltrative
malignant neoplasms, these lesions warrant excision
with a wide margin, similar to treatment of locally
advanced malignancies.
However, there has been inadequate evidence
to suggest association between XGI and gastro-intestinal
malignancies. Of the eight cases of XGI
of the stomach reported in the literature,8 9 10 11 12 13 co-existence
of XGI with gastric cancer was reported
in three cases. Histological examination of these
cases did not support continuity between the
xanthogranuloma and adenocarcinoma. In the other
case of XGI in the terminal ileum reported by Yoon et
al,2 preoperative endoscopy and biopsy showed ulcers
with acute and chronic inflammation only. However,
surgical resection was considered unavoidable in
view of the radiological findings highly suggestive of
appendiceal cancer. While preoperative endoscopic
biopsy may not be helpful to exclude malignancy as
most of the lesions are submucosal, intra-operative
frozen section may be helpful to avoid unnecessary
radical surgery.
Conclusion
We report a case of XGI with full-thickness
involvement of the terminal ileum presenting with
a tender intraperitoneal mass. This report aimed to
emphasise ileal involvement of XGI, although rare,
as one of the differential diagnoses of mass lesions
in the small bowel mimicking malignant neoplasms.
Declaration
No conflicts of interest were declared by the authors.
References
1. Oberling C. Retroperitoneal xanthogranuloma. Am J
Cancer 1935;23:477-89. CrossRef
2. Yoon JS, Jeon YC, Kim TY, et al. Xanthogranulomatous
inflammation in terminal ileum presenting as an
appendiceal mass: case report and review of the literature.
Clin Endosc 2013;46:193-6. CrossRef
3. Lo CY, Lorentz TG, Poon CS. Xanthogranulomatous
inflammation of the sigmoid colon: a case report. Aust N Z
J Surg 1996;66:643-4. CrossRef
4. Oh YH, Seong SS, Jang KS, et al. Xanthogranulomatous
inflammation presenting as a submucosal mass of the
sigmoid colon. Pathol Int 2005;55:440-4. CrossRef
5. Anadol AZ, Gonul II, Tezel E. Xanthogranulomatous
inflammation of the colon: a rare cause of cecal mass with
bleeding. South Med J 2009;102:196-9.
6. Dhawan S, Jain D, Kalhan SK. Xanthogranulomatous
inflammation of ascending colon with mucosal
involvement: report of a first case. J Crohns Colitis
2011;5:245-8. CrossRef
7. Guo G, Greenson JK. Histopathology of interval (delayed)
appendectomy specimens: strong association with
granulomatous and xanthogranulomatous appendicitis.
Am J Surg Pathol 2003;27:1147-51. CrossRef
8. Zhang L, Huang X, Li J. Xanthogranuloma of the stomach:
a case report. Eur J Surg Oncol 1992;18:293-5.
9. Guarino M, Reale D, Micoli G, Tricomi P, Cristofori
E. Xanthogranulomatous gastritis: association with
xanthogranulomatous cholecystitis. J Clin Pathol
1993;46:88-90. CrossRef
10. Lespi PJ. Gastric xanthogranuloma (inflammatory
malignant fibrohistiocytoma). Case report and literature
review [in Spanish]. Acta Gastroenterol Latinoam
1998;28:309-10.
11. Lai HY, Chen JH, Chen CK, et al. Xanthogranulomatous
pseudotumor of stomach induced by perforated peptic
ulcer mimicking a stromal tumor. Eur Radiol 2006;16:2371-2. CrossRef
12. Kubosawa H, Yano K, Oda K, et al. Xanthogranulomatous
gastritis with pseudosarcomatous changes. Pathol Int
2007;57:291-5. CrossRef
13. Kinoshita H, Yamaguchi S, Sakata Y, Arii K, Mori K,
Kodama R. A rare case of xanthogranuloma of the stomach
masquerading as an advanced stage tumor. World J Surg
Oncol 2011;9:67. CrossRef
14. Rogers S, Slater DN, Anderson JA, Parsons MA. Cutaneous
xanthogranulomatous inflammation: a potential indicator
of internal disease. Br J Dermatol 1992;126:290-3.
15. Chuang YF, Cheng TI, Soong TC, Tsou MH.
Xanthogranulomatous appendicitis. J Formos Med Assoc
2005;104:752-4. CrossRef
Find HKMJ in MEDLINE: