Hong Kong Med J 2014;20:258–60 | Number 3, June 2014
DOI: 10.12809/hkmj134025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
A novel mutation in pseudohypoparathyroidism type 1a in a Chinese woman and her son with hypocalcaemia
Vicki HK Tam, FHKCP, FHKAM (Medicine)1;
Sammy PL Chen, MRCP (UK), FHKAM (Pathology)2;
Chloe M Mak, MD, FHKAM (Pathology)2;
LM Fung, FRCP (Edin), FHKAM (Medicine)1;
CY Lee, FRCP (Edin), FHKAM (Paediatrics)3;
Albert YW Chan, MD, FHKAM (Pathology)2
1 Department of Medicine and Geriatrics, Caritas Medical Centre,
Shamshuipo, Hong Kong
2 Kowloon West Cluster Laboratory Genetic Service, Chemical Pathology
Laboratory, Department of Pathology, Princess Margaret Hospital,
Laichikok, Hong Kong
3 Department of Paediatrics and Adolescent Medicine, Caritas Medical
Centre, Shamshuipo, Hong Kong
Corresponding author: Dr Vicki HK Tam (vickitam@gmail.com)

Abstract
Pseudohypoparathyroidism is a rare genetic disorder
characterised by end-organ resistance to parathyroid
hormone due to a defect of the guanine nucleotide–binding protein alpha that simulates activity of the
polypeptide 1 (GNAS) gene. Patients with type 1a
pseudohypoparathyroidism display different features
of Albright’s hereditary osteodystrophy as well as
multi-hormone resistance. We describe a Chinese
woman and her son, who presented with different
symptoms of pseudohypoparathyroidism and
clinically manifested different degree of Albright’s
hereditary osteodystrophy. Genetic study detected
a mutation [NM_000516.4(GNAS):c682C>T
(p.Arg228Cys)] in the GNAS gene.
Introduction
Pseudohypoparathyroidism (PHP) is characterised
by hypocalcaemia and hyperphosphataemia due to
resistance to parathyroid hormone (PTH). This was
the first hormone resistance syndrome described in
1942 by Fuller Albright and his colleagues.1
There are three forms of PHP, namely: PHP-1,
PHP-2, and pseudopseudohypoparathyroidism
(PPHP). Evidently, PHP-1 differs from PHP-2
in that patients with the former show a blunted
urinary cyclic AMP (cAMP) response to exogenous
administration of PTH, whereas those with PHP-2
have normal urinary cAMP excretion but a blunted
phosphaturic response. Moreover, PHP-1 is further
classified into three different subtypes (1a, 1b, and
1c) based on the presence or absence of Albright’s
hereditary osteodystrophy (AHO), which typically
includes short stature, obesity, brachydactyly, ectopic
ossification, and mental retardation. Both PHP-1a
and PHP-1c display features of AHO, but PHP-1b
does not. Furthermore, PHP-1a is distinguished
from PHP-1c in that it contains the inactivating
mutation in the gene encoding Gsα (GNAS).
Patients with both PHP-1a and PPHP carry
heterozygous inactivating GNAS mutations. Apart
from having AHO, patients with PHP-1a show
resistance to hormones that act via G protein–coupled receptors. Patients with PPHP show only
the features of AHO.
Case report
A 44-year-old woman, with no significant past
medical illness, presented to the Department of
Orthopaedics and Traumatology of Caritas Medical
Centre in 2006 because of progressive weakness and
numbness in both lower limbs. These symptoms had
been present for years and she was only recently
unable to walk. Magnetic resonance imaging of the
cervical spine revealed osteophytosis and thickening
of posterior longitudinal ligament, resulting in
narrowing of the spinal canal at multiple levels with
compression on the cervical cord. She was diagnosed
as having cervical spondylosis, and laminoplasty was
performed in September 2007. Postoperatively, she
was noted to have hypocalcaemia with a total serum
calcium level of 1.81 mmol/L (reference range [RR],
2.10-2.60 mmol/L), and a serum phosphate level of
1.08 mmol/L (RR, 0.8-1.5 mmol/L). The serum PTH
level was 258 pg/mL (RR, 11-54 pg/mL); her adjusted
calcium level was 2.12 mmol/L.
The patient was brought up by her stepmother
since she was young. She got divorced and
had no siblings and lost contact with her biological
parents and their families. She also had a history
of oligomenorrhoea since menarche with only one
to two menstrual periods per year. On physical
examination, her body weight was 87.2 kg and height
1.61 m, with a body mass index of 33.6 kg/m2. She was
obese with a moon face, but there was no definite brachydactyly. The clinical diagnosis was PHP.
Other investigations revealed that she had
subclinical hypothyroidism with a serum thyroid-stimulating
hormone (TSH) level of 7.64 mIU/L
(RR, 0.50-4.70 mIU/L) and serum free T4 level
of 10.7 pmol/L (RR, 9.1-23.8 pmol/L). The anti-thyroglobulin
antibody titre was < 1/100 but the
anti-microsomal antibody titre was 1/24 600. Her
serum follicle-stimulating hormone level was 20 U/L,
serum luteinising hormone level was 14.8 U/L, and
serum oestradiol level was <73 pmol/L. The short
synacthen test (using 250 µg tetracosactrin) showed
a baseline serum cortisol level of 77 nmol/L and peak
level of 500 nmol/L. She was started on calcitriol
and calcium supplement as well as thyroxine
replacement.
She only had one child, a 16-year-old son who
also had “calcium problem”. He was followed up
by the Department of Paediatrics and Adolescent
Medicine of our hospital. In 2003, he had presented
with a generalised tonic-clonic convulsion at the age
of 12 years. At that time, his serum calcium level was
1.46 mmol/L, phosphate level of 1.98 mmol/L, and a
PTH level of 70 pg/mL. The thyroid function test was
normal. He was obese and tall with a body weight of
60 kg (at 97th percentile in the growth chart) and
height of 166.3 cm (>97th percentile). He suffered
from mild mental retardation and studied in special
school. There was mild shortening of the fourth and
fifth metacarpals (Fig). He was diagnosed as having
PHP with AHO features, and received calcitriol and
calcium supplement. Over the years, he had gone
through a normal puberty. The brachydactyly had
become more prominent.
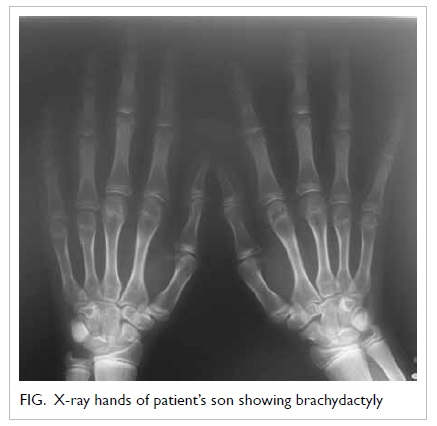
Figure. X-ray hands of patient’s son showing brachydactyly
We performed a mutation analysis on the
GNAS gene of both the mother and her son.
Genomic DNA of both patients was extracted from
peripheral blood leukocytes using the QIAamp Blood Kit (Qiagen, Hilden, Germany). The coding
exons and the flanking regions of the SPG4 gene were
amplified using a polymerase chain reaction and
sequenced. The numbering of nucleotides was based
on GenBank accession number NM_000516.4 with
394 amino acids. Protocol is available on request.
A heterozygous missense mutation,
NM_000516.4(GNAS):c.682C>T (p.Arg228Cys), in
the GNAS gene was identified in both the proband
and her son. This was a novel mutation, with
involvement of a structurally non-conservative
substitution of the evolutionary conserved amino
acid change predicted to affect protein function
by Sorting Intolerant From Tolerant analysis,
Polyphen-2 and MutationTaster analyses. Screening
in 300 normal chromosomes did not suggest this as
a polymorphic site. The diagnosis of the mother was
PHP-1a with mild AHO, and of her son was PHP-1a
with AHO.
Discussion
It appears that PHP-1a is an autosomal dominant
disease in which full clinical and metabolic
abnormalities may not be present initially, but
become apparent later. Patients with PHP-1a showed
a heterozygous inactivating germline mutation
in GNAS, the gene encoding the α-subunit of the
stimulatory GTP binding protein (Gsα). This could
lead to a reduced Gsα protein level and cellular
activity and thus the clinical resistance phenotype.2
The GNAS gene maps to 20q13 and contains
13 exons.3 The mutation can be localised in the
entire coding region of the gene. All exons can be
affected by loss-of-function alterations with the
exception of exon 3, where no mutations have
been detected to date.4 The hot-spot mutations
accounting for about 20% of all mutations so far
described have been identified on exon 7.5 As for the
types of mutations, small insertions, deletions and
amino-acid substitutions predominate. Our patient
showed a novel heterozygous missense mutation of
exon 9 in the GNAS gene, which is considered to be functionally deleterious.
Maternal inheritance of this GNAS mutation
leads to PHP-1a (ie AHO plus hormone resistance),
while paternal inheritance of the same mutation
leads to PPHP (ie AHO only).6 This imprinted
mode of inheritance for hormone resistance can be
explained by the predominantly maternal expression
of Gsα in certain tissues, including renal proximal
tubules.7 In our case, the son inherited the same
molecular defect from his mother, resulting in
PHP-1a. It could be postulated that the proband
should also inherit the condition from her mother,
but this remains to be substantiated as there is not
much helpful information available.
Our patient (the mother) also had subclinical
hypothyroidism; the anti-microsomal antibody
was positive. She had evidence of hypogonadism
(oligomenorrhoea and low oestradiol levels). By
contrast, her son had normal thyroid function, and he
had a normal puberty. Co-existing endocrinological
abnormalities may ensue, as individuals with PHP-1a
also demonstrate resistance to other hormones
such as TSH, gonadotropins, and growth hormone-releasing
hormone. Fernandez-Rebollo et al8 showed
that most patients with PHP-1a have TSH resistance,
which is usually mild, and manifests during
childhood or adolescence. Goitre and anti-thyroid
antibodies are usually absent.9 Clinical evidence of
hypogonadism is common in PHP-1a, particularly in
females, and manifests as delayed sexual maturation,
amenorrhoea, oligomenorrhoea, and/or infertility.
Affected individuals usually have slight hypo-estrogenism,
but no definite evidence of increased
basal or gonadotropin-releasing hormone–stimulated levels of circulating gonadotropins.10
Mantovani and Spada11 demonstrated that growth
hormone deficiency is also common in patients with
PHP-1a. However, the relevance of growth hormone
deficiency on final height and obesity in these patients
is not certain, because PPHP patients (who do not
have hormonal resistance) also have short statures
together with obesity. That study also evaluated the
adrenocortical and corticotropin responsiveness
in patients with PHP-1a; all of whom showed a
normal response to 1 µg adrenocorticotropin and
to corticotropin-releasing factor. Normal pituitary-adrenal
function in these patients suggested that
the presence of Gsα imprinting within the pituitary
gland is cell-type specific.12
Spinal cord compression is a rare neurological
complication of PHP or PPHP, which is due to
ossification of the posterior longitudinal ligament
that may compress the spinal cord.13 Presentations
include spastic paraparesis, tetraparesis, and urinary
incontinence.14 Many such patients endure long-term
disability despite neurosurgical intervention.
Our patient had a long history of neurological symptoms but presented late. After laminoplasty,
she still had residual weakness. Hence, spinal cord
compression should also be considered in patients
with PHP or PPHP who present with neurological
symptoms.
We have demonstrated a novel mutation in the
GNAS gene in a small Chinese family with PHP-1a.
One family member presented with spinal cord
compression, which is a rare complication in PHP
caused by ectopic ossification. Moreover, associated
endocrinopathies, especially hypothyroidism and
hypogonadism, are common in PHP-1a.
References
1. Albright F, Burnett CH, Smith PH, Parson W. Pseudohypoparathyroidism—an example of ‘Seabright-Bantam syndrome’. Endocrinology 1942;30:922-32.
2. Thakker RV. Genetic developments in hypoparathyroidism. Lancet 2001;357:974-6. CrossRef
3. Pattern JL, Johns DR, Valle D, et al. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. N Engl J Med 1990;322:1412-9. CrossRef
4. Mantovani G, Spada A. Mutations in the Gs alpha gene causing hormone resistance. Best Pract Res Clin Endocrinol Metab 2006;20:501-13. CrossRef
5. Yu S, Yu D, Hainline BE, et al. A deletion hot-spot in exon 7 of the Gs alpha gene (GNAS1) in patients with Albright hereditary osteodystrophy. Hum Mol Genet 1995;4:2001-2. CrossRef
6. Bastepe M. The GNAS locus and pseudohypoparathyroidism. Adv Exp Med Biol 2008;626:27-40. CrossRef
7. Bastepe M, Jüppner H. Pseudohypoparathyroidism. New insights into an old disease. Endocrinol Metab Clin North Am 2000;29:569-89. CrossRef
8. Fernandez-Rebollo E, Barrio R, Pérez-Nanclares G, et al. New mutation type in pseudohypoparathyroidism type 1a. Clin Endocrinol (Oxf) 2008;69:705-12. CrossRef
9. Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 2001;22:675-705.
10. Namnoum AB, Marriam GR, Moses AM, Levine MA. Reproductive dysfunction in women with Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab 1998;83:824-9.
11. Mantovani G, Spada A. Resistance to growth hormone releasing hormone and gonadotropins in Albright’s hereditary osteodystrophy. J Paediatr Endocrinol Metab 2006;19:663-70. CrossRef
12. Mantovani G, Maghnie M, Weber G, et al. Growth hormone-releasing hormone resistance in pseudohypoparathyroidism type Ia: new evidence of imprinting for the Gs alpha gene. J Clin Endocrinol Metab 2003;88:4070-4. CrossRef
13. Iwase T, Nokura K, Mizuno T, Inagaki T. Spastic tetraparesis in a patient with pseudopseudohypoparathyroidism. J Neurol 2002;249:1457-8.
14. Alam SM, Kelly W. Spinal cord compression associated with pseudohypoparathyroidism. J R Soc Med 1990;83:50-1.