DOI: 10.12809/hkmj144237
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Unexplained childhood anaemia: idiopathic pulmonary hemosiderosis
KK Siu, MB, ChB, MRCPCH1; Rever Li, MB, ChB, FHKAM (Paediatrics)2; SY Lam, MB, BS, FHKAM (Paediatrics)2
1 Department of Paediatrics, Kwong Wah Hospital, Yaumatei, Hong Kong
2 Department of Paediatrics, Tuen Mun Hospital, Tuen Mun, Hong Kong
Corresponding author: Dr KK Siu (skk053@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
This report demonstrates pulmonary haemorrhage
as a differential cause of anaemia. Idiopathic
pulmonary hemosiderosis is a rare disease in children;
it is classically described as a triad of haemoptysis,
pulmonary infiltrates on chest radiograph, and iron-deficiency
anaemia. However, anaemia may be the
only presenting feature of idiopathic pulmonary
hemosiderosis in children due to occult pulmonary
haemorrhage. In addition, the serum ferritin is falsely
high in idiopathic pulmonary hemosiderosis which
increases the diagnostic difficulty. We recommend
that pulmonary haemorrhage be suspected in any
child presenting with iron-deficiency anaemia and
persistent bilateral pulmonary infiltrates.
Case report
We report the case of a 5-year-old boy, who
presented with recurrent episodes of unexplained
iron-deficiency anaemia in February 2010 since the
age of 27 months. Serial chest X-rays (CXRs) showed
bilateral reticulonodular haziness. Bronchoalveolar
lavage and lung biopsy confirmed the diagnosis of
idiopathic pulmonary hemosiderosis (IPH).
The patient first presented at 27 months of age
in Mainland China with malaise, loss of appetite,
and shortness of breath for 10 days. He did not have
fever, cough, or haemoptysis. He received two doses
of H1N1 vaccination before pallor was noted. There
was no history of drug or herb intake.
The presenting haemoglobin (Hb) level was
65 g/L (reference range [RR], 115-145 g/L), mean
corpuscular volume (MCV) 84.4 fL (RR, 76-90 fL),
mean corpuscular haemoglobin (MCH) level 25.8 pg (RR,
25-31 pg), and reticulocyte count 10.4% (reference
level [RL], <2%). White cell count and platelet count
were unremarkable. Blood smear showed moderate
anisopoikilocytosis with polychromasia. Bone
marrow aspiration and trephine biopsy revealed
active marrow with erythroid preponderance.
Other investigations were performed for
anaemia. Serum lactate dehydrogenase (LDH) level was
elevated to 980 U/L (RL, <615 U/L). Haptoglobin level
was reduced (<0.09 g/L; RR, 0.3-2.7 g/L). Direct
Coombs test was negative. Bilirubin was normal.
Urine for bilirubin and urobilinogen was negative.
Haemoglobin pattern, serum vitamin B12, and G6PD
activity were unremarkable. Serum ferritin level
was 137 pmol/L (RR, 45-449 pmol/L). Antinuclear
antibody assay was negative. Flow cytometry
showed normal expression of CD55 and CD59 which
ruled out paroxysmal nocturnal haemoglobinuria.
Donath-Landsteiner antibody assay was negative,
which excluded the diagnosis of paroxysmal cold
haemoglobinuria.
Blood work for viral infection including
antibodies to parvovirus, Epstein-Barr virus (EBV),
mycoplasma, and viral titres was unremarkable.
Screening for blood loss was negative. Stool
for occult blood and urine for Hb were negative. Red
blood cell scan showed no evidence of haemorrhage.
Blood transfusion was given to correct the
anaemia. Microcytic hypochromic anaemia was
noted at 2 months after presentation. Haemoglobin
level was 54 g/L. Mean corpuscular volume
dropped from 84.4 fL to 68 fL. Mean corpuscular
haemoglobin level was 21.3 pg. Iron profile showed a low
serum iron level of 3 µmol/L (RR, 5-20 µmol/L), elevated
total iron-binding capacity (TIBC) of 70 µmol/L
(RR, 37-68 µmol/L), and iron saturation of 4% (RR,
20-55%). However, the serum ferritin level was normal at
220 pmol/L (RR, 45-449 pmol/L). Iron supplement
was started as a therapeutic trial for suspected iron-deficiency
anaemia.
His Hb level remained stable in the next 2 years.
However, in the subsequent 8 months, there were
three intermittent episodes of anaemia (lowest Hb level,
60 g/L). The anaemia occurred with fever and cough
which were considered symptoms of pneumonia
and upper respiratory tract infection. There was no
history of haemoptysis.
At 36 months after the presentation, he was
admitted again for pallor, fatigue, and fever. This time,
he needed oxygen therapy. Chest was clear with mild
subcostal insucking. Hepatomegaly of 4 cm below the
costal margin was noted. Chest X-ray showed bilateral
reticulonodular haziness (Fig 1). Haemoglobin level was 61 g/L. Both MCV (78 fL) and MCH level (25 pg) were
in the lower normal limits. The serum ferritin level was
879 pmol/L. A diagnosis of pneumonia was made;
he was started on oral co-amoxiclav (amoxicillin and
clavulanic acid) and azithromycin and given blood
transfusion. Fever subsided and oxygen was weaned
off. Ultrasonography of abdomen performed 1
month later showed no hepatomegaly; mild hepatic
coarsening was suggestive of parenchymal disease.
Liver function tests were normal all along. However,
the patient tested positive for EBV immunoglobulin
(Ig) M antibodies. Thus, he was diagnosed to have
pneumonia with EBV infection.
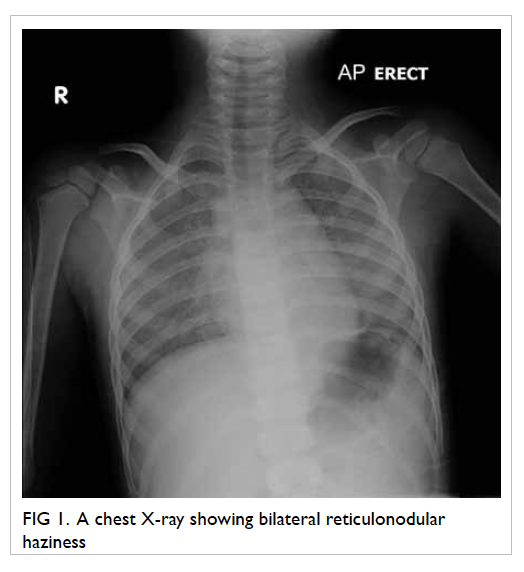
Figure 1. A chest X-ray showing bilateral reticulonodular haziness
Review of old CXRs showed similar
reticulonodular shadows. In view of his history of
recurrent iron-deficiency anaemia and CXR findings,
pulmonary hemosiderosis was suspected. Flexible
bronchoscopy was performed; bronchoscopic lavage
over left lingular and right middle lobe showed
blood-stained fluid. Bronchoalveolar lavage yielded
abundant hemosiderin-laden macrophages (HLM
index, 92%). High-resolution computed tomography
of thorax showed extensive ground glass opacities
and reticular shadows, suggestive of interstitial lung
disease. Diffuse visceral pleural brownish deposits
were noted over the entire left lung on video-assisted
thoracoscopy. Lung biopsy was performed to rule
out systemic disorders with pulmonary capillaritis,
which could cause diffuse alveolar haemorrhage
(DAH). These included Goodpasture’s syndrome, IgA
nephropathy, Wegener’s granulomatosis, systemic
lupus erythematosus, and antiphospholipid syndrome.
Biopsy of lung tissue showed numerous HLM. There
was no evidence of capillaritis or vasculitis. Absence
of fibrosis and negative exposure history also excluded
hypersensitivity pneumonitis. Immunostaining for
IgG, IgM, and IgA was negative. The overall picture
was compatible with pulmonary hemosiderosis.
Further blood investigations were performed to
exclude systemic causes of pulmonary haemorrhage
as stated above. Antiglomerular basement membrane
antibodies, rheumatoid factor, anti-neutrophil
cytoplasmic antibodies, antinuclear antibodies, anti-extractable
nuclear antibodies, and anti-cardiolipin
IgG antibodies were not detected. Furthermore,
the patient was negative for anti-transglutaminase
antibody for coeliac disease. He tested weakly
positive for IgE antibodies against cow’s milk.
Immunoglobulin pattern was unremarkable apart
from mildly raised IgA antibody level at 2.28 g/L
(range, 0.5-1.92 g/L). Renal function and urinalysis
were normal.
A diagnosis of IPH was made based on the
above findings. Oral prednisolone was started
after the diagnosis for disease control. Chest X-ray
performed after 3 months of prednisolone showed
improvement in reticulonodular densities (Fig 2). We
plan to monitor the disease with clinical symptoms,
Hb levels, LDH levels, CXRs, and spirometry.
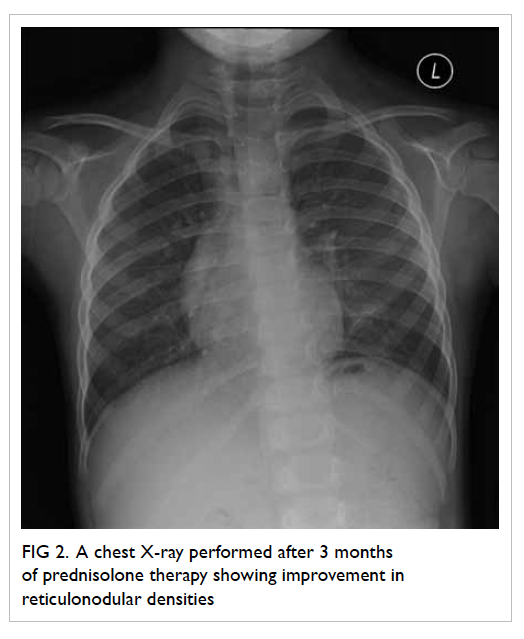
Figure 2. A chest X-ray performed after 3 months of prednisolone therapy showing improvement in reticulonodular densities
Discussion
Idiopathic pulmonary hemosiderosis is a rare disease
in children with an unknown aetiology. The estimated
yearly incidence among Swedish children from 1960
through 1979 was 0.24 per 1 000 000 children.1 A
retrospective review of records from a tertiary
paediatric hospital in northern Taiwan noted five
cases over 25 years.2 Patients classically presented
with a triad of recurrent or chronic pulmonary
symptoms (cough, dyspnoea, wheeze, haemoptysis),
pulmonary infiltrates on CXR, and iron-deficiency
anaemia. Our patient had only anaemia without
obvious underlying causes. Subsequent CXR changes
led to the suspicion of IPH.
Serum ferritin has been traditionally taken
as a reliable surrogate marker of body iron stores.
Hypoferritinaemia is commonly used as a diagnostic
marker for iron deficiency.3 However, as it is an acute-phase reactant, abnormally raised serum ferritin level
may be seen during acute infection or liver disease
even in the presence of iron deficiency.4 In IPH,
iron study usually shows low serum iron with low
iron saturation, and microcytosis and hypochromia
in the blood picture. However, plasma ferritin level
can be normal or elevated in IPH because of alveolar
synthesis and release into the circulation and does
not reflect the iron deposits in the body.5 This makes
the diagnosis of iron-deficiency anaemia in IPH
difficult. We recommend the use of serum iron and
transferrin saturation (serum iron/TIBC) instead to
evaluate suspected iron-deficiency anaemia.4
Diagnosis of IPH is based on exclusion of other
causes of intrapulmonary haemorrhage and systemic
diseases. In the absence of systemic disease, findings
of HLM in bronchoscopic lavage or gastric aspirate/sputum along with chronic pulmonary symptoms
lead to a diagnosis of IPH. Lung biopsy is the gold
standard for diagnosis. We performed lung biopsy to
exclude pulmonary capillaritis, which is one of the
causes of DAH. Pulmonary capillaritis is a small-vessel
vasculitis, which can occur as an isolated
condition or in association with multiple systemic
vasculitides. Isolated DAH without identifiable
causation or associated disease is referred to as IPH.6
Daily oral corticosteroids or weekly
intravenous pulse methylprednisolone is commonly
used in the induction treatment of IPH. Other
immunosuppressive agents such as azathioprine,
cyclophosphamide, and hydroxychloroquine have
also been used alone or in combination with oral
corticosteroids.7 8 9 10 11 Low-dose oral corticosteroids,
azathioprine, or methotrexate are used in
maintenance phase. As there is lack of large patient
series and inadequate follow-up in previous studies,
the prognosis of IPH remains unclear. However,
aggressive treatment with the use of corticosteroids
and immunosuppressive agents are associated with
a prolonged survival and improved prognosis.12
Long-term low-dose corticosteroid therapy was also
reported to result in a milder disease course and
prevent bleeding crisis.13
In conclusion, iron-deficiency anaemia results
from poor dietary intake of iron in infants and
toddlers. However, every child older than 24 months
presenting with iron-deficiency anaemia should be
evaluated for chronic blood loss. In this report, we
have illustrated that anaemia without any respiratory
symptoms can be the sole presenting feature of IPH,
preceding other signs and symptoms, especially in
young children. Haemoptysis may not be present in
young children with IPH, as they tend to swallow
their sputum. We recommend that when children
present with unexplained anaemia and bilateral lung
infiltrations, pulmonary haemorrhage should be
suspected.
References
1. Kjellman B, Elinder G, Garwicz S, Svan H. Idiopathic
pulmonary haemosiderosis in Swedish children. Acta
Paediatr Scand 1984;73:584-8. Crossref
2. Yao TC, Hung IJ, Wong KS, Huang JL, Niu CK. Idiopathic
pulmonary haemosiderosis: an Oriental experience. J
Paediatr Child Health 2003;39:27-30. Crossref
3. Rybo E. Diagnosis of iron deficiency. Scand J Haematol
Suppl 1985;43:5-39.
4. Li CH, Lee AC, Mak TW, Szeto SC. Transferrin saturation
for the diagnosis of iron deficiency in febrile anaemic
children. Hong Kong Pract 2003;25:363-6.
5. Ioachimescu OC, Sieber S, Kotch A. Idiopathic pulmonary
haemosiderosis revisited. Eur Respir J 2004;24:162-70. Crossref
6. Fullmer JJ, Langston C, Dishop MK, Fan LL. Pulmonary
capillaritis in children: a review of eight cases with
comparison to other alveolar hemorrhage syndromes. J
Pediatr 2005;146:376-81. Crossref
7. Milman N, Pedersen FM. Idiopathic pulmonary
haemosiderosis. Epidemiology, pathogenic aspects and
diagnosis. Respir Med 1998;92:902-7. Crossref
8. Rossi GA, Balzano E, Battistini E, et al. Long-term
prednisolone and azathioprine treatment of a patient with
idiopathic pulmonary hemosiderosis. Pediatr Pulmonol
1992;13:176-80. Crossref
9. Colombo JL, Stolz SM. Treatment of life-threatening
primary pulmonary hemosiderosis with cyclophosphamide.
Chest 1992;102:959-60. Crossref
10. Zaki M, al Saleh Q, al Mutari G. Effectiveness of chloroquine
therapy in idiopathic pulmonary hemosiderosis. Pediatr
Pulmonol 1995;20:125-6. Crossref
11. Bush A, Sheppard MN, Warner JO. Chloroquine in
idiopathic pulmonary haemosiderosis. Arch Dis Child
1992;67:625-7. Crossref
12. Saeed MM, Woo MS, MacLaughlin EF, Margetis MF,
Keens TG. Prognosis in pediatric idiopathic pulmonary
hemosiderosis. Chest 1999;116:721-5. Crossref
13. Kiper N, Göçmen A, Ozçelik U, Dilber E, Anadol D. Long-term
clinical course of patients with idiopathic pulmonary
hemosiderosis (1979-1994): prolonged survival with low-dose
corticosteroid therapy. Pediatr Pulmonol 1999;27:180-4. Crossref