DOI: 10.12809/hkmj187403
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
COMMENTARY
Paraneoplastic neuromyelitis optica spectrum disorder
diagnosed with mantle cell lymphoma
Samuel SY Wang, BMed1; Thomas KH Lau,
MB, BS 2
1 Faculty of Medicine, University of New
South Wales, Kensington, Australia
2 Department of Clinical Oncology,
Prince of Wales Hospital, Shatin, Hong Kong
Corresponding author: (lkh195@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Paraneoplastic neurological syndromes (PNS) are
well-recognised neurological symptoms that occur at increased frequency in
cancer patients. Paraneoplastic neurological syndromes are independent of
metastasis, direct tumour infiltration, or known indirect mechanisms such
as toxicity, ectopic hormone secretion, or induced coagulopathies.1 Paraneoplastic neurological syndromes may affect any
part of the nervous system, and are believed to result when an immunologic
response is directed against shared antigens that are ectopically
expressed by the tumour, but otherwise predominantly expressed by the
nervous system (onconeural antigens).1
Antibodies can be detected in the serum or cerebrospinal fluid of many,
but not all, patients with PNS.2
Diagnosing PNS requires identification of the type of neurological
syndrome based on neurological signs and symptoms, the detection of
well-characterised onconeural antibodies, and the presence of a cancer.3
Paraneoplastic neurological syndromes are rare in
patients with solid tumours, and probably even rarer among patients with
lymphomas.4 The predominant types
of PNS in lymphomas are paraneoplastic cerebellar degeneration in
Hodgkin’s lymphoma, and dermatomyositis/polymyositis in both Hodgkin’s
lymphoma and non–Hodgkin’s lymphoma. Other PNS are uncommon and are
reported only in isolated case reports and case series.2
Neuromyelitis optica spectrum disorder (NMOSD) is
an inflammatory disorder of the central nervous system characterised by
severe, immune-mediated demyelination, and axonal damage predominantly
targeting optic nerves and spinal cord. Traditionally considered a variant
of multiple sclerosis, presence of the disease-specific aquaporin-4
antibody, which plays a direct role in the pathogenesis of NMOSD,
distinguishes the two entities.5
Recently, NMOSD is increasingly recognised as a
paraneoplastic disorder especially in men or in patients who present in
older age.6 Paraneoplastic NMOSD
has been reported in a wide variety of tumour histological types but
mostly in solid tumours.6
We recently encountered a definite case of PNS in a
patient who was diagnosed with mantle cell lymphoma (MCL) and shortly
afterwards developed neurological symptoms due to NMOSD. An 83-year-old
Chinese man presented to Prince of Wales Hospital, Hong Kong with change
in bowel habit and per-rectal bleeding since January 2015. An
oesophageal-gastro-duodenoscopy showed diffuse gastritis and biopsy
confirmed infiltration by MCL. Subsequent colonoscopy also showed
involvement of the rectum by MCL. In March 2015, the patient then
presented to Alice Ho Miu Ling Nethersole Hospital, Hong Kong with
acute-onset left-sided numbness and right-sided hemiparesis. Brain
computed tomography was normal. Subsequent magnetic resonance imaging
spine revealed a hyperintense signal from C3 to C8 with oedema in the
spinal cord without an intramedullary space-occupying lesion (Fig
a). He was treated with pulse methylprednisolone for the transverse
myelitis which showed gradual neurological improvement. A lumbar puncture
was then performed which showed normal total protein and glucose, negative
white cell and cytology; however, the sample was obtained after one dose
of pulse methylprednisolone therapy. Subsequent staging positron emission
tomography/computed tomography was performed and showed hypermetabolic
uptakes at the gastric pylorus, rectosigmoid colon, and spleen. Peripheral
blood and bone marrow examination confirmed involvements by MCL. In view
of the patient’s advanced age and borderline performance status, he was
treated with rituximab, chlorambucil, and prednisolone combination
therapy. After one cycle of treatment, he developed an acute onset of
right eye vision loss, while his right hemiparesis showed persistent
improvement. Magnetic resonance imaging of the brain and orbit was then
performed, which showed right optic neuritis (Fig b). He was treated with another course of pulse
methylprednisolone. Serum anti–aquaporin-4 antibody tested positive, and
thus neurologists diagnosed NMOSD. He received a total of six courses of
rituximab, chlorambucil, and prednisolone combination therapy with gradual
partial improvement of right eye vision, almost full recovery of limb
power, and cessation of per-rectal bleeding. The patient was thus offered
maintenance rituximab therapy. The patient remained stable until 16 months
later when he developed per-rectal bleeding and palatal ulcers. Biopsy of
the palatal ulcers revealed MCL involvement, and MCL therapy was switched
to rituximab-cyclophosphamide-vincristine-prednisolone for six cycles. The
lymphoma progressed on treatment with recurrence of palate nodules,
parotid masses, and weakening limb power. Lenalidomide therapy was offered
but was not started as the drug was a self-financed item and the patient
could not afford it. He was started on maintenance azathioprine for
prevention of relapse of NMOSD by the neurologists. However, the patient
later developed an acute left vision loss due to left optic neuritis, and
sustained a fall injury which resulted in a right hip fracture. The
patient became bed-ridden despite hip fracture fixation and was deemed
unfit for further anti-cancer therapy. He was then placed on best
supportive care.
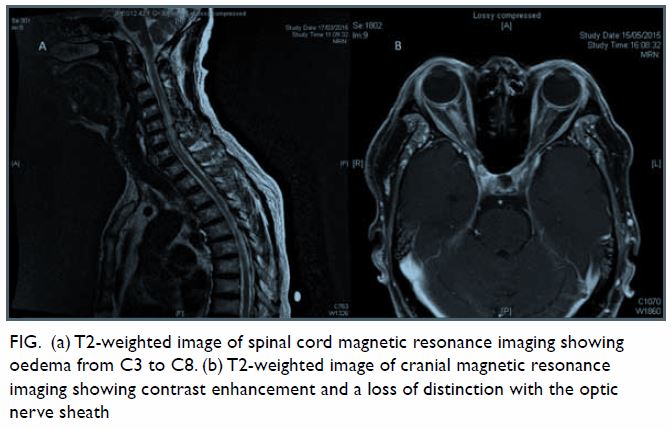
Figure. (a) T2-weighted image of spinal cord magnetic resonance imaging showing oedema from C3 to C8. (b) T2-weighted image of cranial magnetic resonance imaging showing contrast enhancement and a loss of distinction with the optic nerve sheath
Previous reported cases of paraneoplastic NMOSD are
mostly associated with solid cancers, most commonly breast and lung
cancers. To the best of our knowledge, this is the first reported case of
MCL to be associated with paraneoplastic NMOSD. This unusual case supports
the suggestion that for patients with NMOSD who present in older age, as
opposed to third or fourth decade in idio-pathic cases, an underlying
malignancy should be suspected. This case also illustrates that effective
treatment of the underlying lymphoma is important in controlling
neurological disease, though the use of expensive drugs can be challenging
in a resource-poor setting. Our study highlights the importance of the
correct diagnosis of PNS. Early recognition of a neurological syndrome as
paraneoplastic often leads to the discovery and treatment of the
underlying tumour, which is a crucial step in the management of the PNS.1 2
Author contributions
All authors have made substantial contributions to
the concept or design; acquisition of data; analysis or interpretation of
data; drafting of the article; and critical revision for important
intellectual content.
Funding/support
This article received no specific grant from any
funding agency in the public, commercial, or not-for-profit sectors.
Declaration
All authors have disclosed no conflicts of
interest. All authors had full access to the data, contributed to the
study, approved the final version for publication, and take responsibility
for its accuracy and integrity.
References
1. Darnell RB, Posner JB. Oxford, UK:
Oxford University Press; 2011. Paraneoplastic syndromes. Contemporary
Neurology Series 79: 1-482.
2. Graus F, Ariño H, Dalmau J.
Paraneoplastic neurological syndromes in Hodgkin and non-Hodgkin
lymphomas. Blood 2014;123:3230-8. Crossref
3. Graus F, Delattre JY, Antoine JC, et al.
Recommended diagnostic criteria for paraneoplastic neurological syndromes.
J Neurol Neurosurg Psychiatry 2004;75:1135-40. Crossref
4. Giometto B, Grisold W, Vitaliani R, et
al. Paraneoplastic neurologic syndrome in the PNS euronetwork database: a
European study from 20 centers. Arch Neurol 2010;67:330-5. Crossref
5. Papadopoulos MC, Verkman AS. Aquaporin 4
and neuromyelitis optica. Lancet Neurol 2012;11:535-44. Crossref
6. Sepulveda M, Sola-Valls N, Escudero D,
et al. Clinical profile of patients with paraneoplastic neuromyelitis
optica spectrum disorder and aquaporin-4 antibodies. Mult Scler
2017:1352458517731914. Crossref