Hong
Kong Med J 2018 Dec;24(6):561–70 | Epub 3 Dec 2018
DOI: 10.12809/hkmj187487
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Clinical and genetic profile of congenital long QT
syndrome in Hong Kong: a 20-year experience in
paediatrics
SY Kwok, MB, ChB, FHKAM (Paediatrics)1;
Anthony PY Liu, MB, BS, FHKAM (Paediatrics)2;
Cindy YY Chan, BSc1;
KS Lun, MB, BS, FHKAM (Paediatrics)1;
Jasmine LF Fung, BBiomedSc2;
Christopher CY Mak, MB, ChB2;
Brian HY Chung, MB, BS, FHKAM (Paediatrics)2;
TC Yung, MB, BS, FHKAM (Paediatrics)1
1 Department of Paediatric Cardiology, Queen Mary Hospital, Pokfulam,
Hong Kong
2 Department of Paediatrics and Adolescent Medicine, Li Ka Shing Faculty
of Medicine, The University of Hong Kong, Pokfulam, Hong Kong
Corresponding author: Dr Brian HY Chung (bhychung@hku.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Introduction: Congenital long QT syndrome
(LQTS) is a genetically transmitted cardiac
channelopathy that can lead to sudden cardiac death.
This study aimed to report the clinical and genetic
characteristics of all young patients diagnosed with
LQTS in the only tertiary paediatric cardiology
centre in Hong Kong.
Methods: This is a retrospective review of all
paediatric and young adult patients diagnosed at our
centre with LQTS from January 1997 to December
2016. The diagnosis of LQTS was established with
a corrected QT interval (QTc) ≥480 ms, Schwartz
score of >3 points, or the presence of a pathogenic
mutation.
Results: Fifty-nine patients (33 males) from 52
families were included, with a mean age of 8.17
years (range, 0.00-16.95 years) at presentation. Five
patients had concomitant congenital heart diseases.
The mean follow-up duration was 5.33 ± 4.65 years.
The mean QTc in the cohort was 504 ± 47 ms. They
presented with syncope and convulsion (49%),
cardiac arrest (10%), bradycardia and neonatal
atrioventricular block (12%). Fifteen (25%) patients
were asymptomatic at diagnosis. Thirty-eight
(64.4%) patients were confirmed to have a pathogenic
mutation for LQTS genes. Forty-five (76.3%)
patients received beta blocker therapy. Thirteen
(22.0%) patients required implantable cardioverter
defibrillator. There was no mortality in the study
period. The 1-, 5-, and 10-year breakthrough cardiac
event–free rates were 93.0%, 80.7%, and 72.6%,
respectively.
Conclusion: Identification of the disorder,
administration of beta blockers, and lifestyle
modification can prevent subsequent cardiac events
in LQTS. Genotyping in patients with LQTS is
essential in guiding medical therapy and improving
prognosis.
New knowledge added by this study
- Two-thirds of young long QT syndrome patients in Hong Kong carry pathogenic mutations. Concomitant congenital heart disease is present in 8.5% of these patients.
- The current treatment strategy for young long QT syndrome patients in Hong Kong includes lifestyle modification, beta blocker therapy, implantation of a cardioverter defibrillator, and sympathectomy.
- Young long QT syndrome patients in the present study have good prognosis. No mortality was reported in the medium-term follow-up.
- Genetic testing should be performed for all patients with clinical diagnosis of long QT syndrome, to facilitate timely genotype-guided therapy and early detection of affected family members.
- The diagnosis of long QT syndrome should be considered in young patients presenting with syncope and convulsions, as well as those with bradycardia and atrioventricular block in early infancy.
- Sudden cardiac death associated with long QT syndrome is preventable. Facilities for genetic testing and inherited arrhythmia assessment are recommended.
Introduction
Congenital long QT syndrome (LQTS) is a life-threatening
cardiac arrhythmia syndrome, which leads to sudden death in young people.1 Congenital
LQTS is characterised by prolonged QT interval
(QTc) on electrocardiogram (ECG) and occurrence
of syncope or cardiac arrest. During the past two
decades, there have been major advancements in
the understanding of the genetic factors underlying
the clinical manifestations, prognosis, and subtype-specific
therapy of LQTS.1 2 3Seventeen disease-causing
LQTS genes have been identified, each of
which can lead to dysfunction in the potassium,
calcium, or sodium cardiac ion channels.4 In this
study, we review the clinical characteristics, genetic
profile, management strategy, and outcome of our
local LQTS paediatric patients.
Methods
Study population
Our study included all children, adolescents, and
young adults diagnosed with congenital LQTS from
January 1997 to December 2016 in the Department
of Paediatric Cardiology, Queen Mary Hospital,
which is the only tertiary paediatric cardiology
referral centre in Hong Kong. All except three
patients were Chinese. Diagnosis of congenital LQTS
was reviewed with reference to the 2015 European
Society of Cardiology guidelines for the management
of patients with ventricular arrhythmias and the
prevention of sudden cardiac death.5 Long QT
syndrome was diagnosed with either corrected QTc
≥480 ms in repeated 12-lead ECG measurements or
LQTS risk score >3 points, as proposed by Schwartz
et al.6 The presence of a confirmed pathogenic LQTS
mutation, irrespective of the QTc, was also used for
diagnosis of LQTS. Secondary causes of LQTS were
excluded.
Demographic data, clinical presentation,
family history, QTc at presentation, and genetic tests
were retrospectively reviewed. Clinically important
presentation was defined as clinical symptoms
or rhythm disturbances that warranted concern.
Incidental findings of isolated prolonged QTc were
not regarded as clinically important presentation.
We reviewed the treatment modalities,
including beta blocker therapy, implantable
cardioverter defibrillator (ICD), permanent
pacemaker, and left cardiac sympathetic denervation.
Clinical outcomes up to December 2016 were
summarised. Syncope, seizure, aborted cardiac
arrest, appropriate ICD shock, or sudden cardiac
death after diagnosis of LQTS were considered as
breakthrough cardiac events.
Genetic test
Genetic tests were offered to all patients after
informed consent was provided by their parents
or guardians. Blood samples were sent to the
Molecular Genetics Laboratory of Victorian Clinical
Genetic Services, Melbourne, Australia, for genetic
testing. Before 2014, six common LQTS genes were
tested (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2,
KCNJ2) by sequencing of the entire coding region
of all known transcripts of the genes. Multiplex
ligation-dependent probe amplification analysis was
performed on five genes (KCNQ1, KCNH2, SCN5A,
KCNE1, KCNE2) to detect deletions or duplications.
After 2014, next-generation sequencing was used to
identify mutations in an arrhythmia gene panel to
replace sequencing of the six LQTS genes (details
of the arrhythmia full panel can be found at http://www.vcgs.org.au/tests/cardiac-gene-panels). Before
referral to our unit, 13 patients had genetic tests
performed by local or overseas genetic testing
centres.
Mutations in the LQTS loci classified as
pathogenic or likely pathogenic were considered as
genotype positive in our cohort. Cascade testing was
offered to first-degree relatives of patients identified
as genotype positive.
Statistical analysis
Statistical analysis was performed with the SPSS
(Window version 17.0; SPSS Inc, Chicago [IL], United
States). Continuous variables were expressed as mean
± standard deviation, median and range. We used the
standard t test for comparisons of continuous data.
The P<0.05 were deemed statistically significant.
Kaplan-Meier survival curves were created with
censoring at first breakthrough cardiac event or last
follow-up, and analysis was made using the log rank
test.
Results
Demographics and clinical characteristics
During the study period, 59 patients (33 males)
in 52 families were identified who fulfilled the
diagnostic criteria as described. Nine individuals
were diagnosed by ECG screening of our index
cases including a 25-year-old young adult. The mean
follow-up duration of the cohort was 5.33 ± 4.65
years. Four patients were lost to follow-up, but no
death was reported in the territory-wide Hospital
Authority electronic patient record. Five patients
were under the care of adult cardiologists at the
follow-up.
Table 1 shows the characteristics of our
patients. The mean age at diagnosis was 8.12 ± 5.19
years (range, 0-25 years). For those patients who had
clinically important presentation, the mean age at
presentation was 8.75 ± 5.12 years (range, 0.00-16.95
years). Boys were younger than girls at presentation
(6.39 ± 5.00 years vs 10.51 ± 4.55 years, P=0.016).
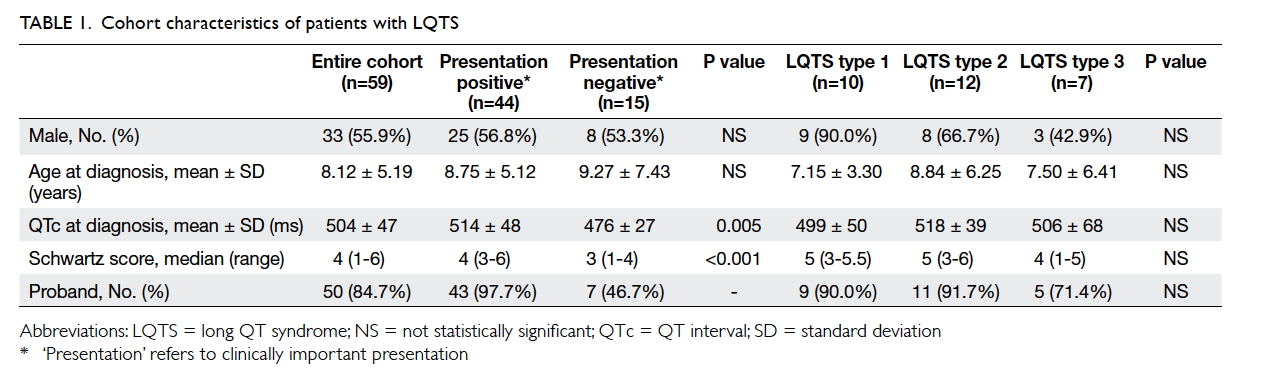
Table 1. Cohort characteristics of patients with LQTS
The mean QTc of the cohort was 504 ± 47 ms.
The median Schwartz score was 4 points (range,
1-6 points). Index patients had longer mean QTc
(512 ± 46 ms) when compared with screened family
members (462 ± 25 ms, P=0.002).
Five (8.47%) patients had congenital heart
defects: secundum atrial septal defect (n=1; genotype
negative), ventricular septal defect (n=1; LQTS type
2 [LQT2]), tetralogy of Fallot (n=2; LQT2 and LQTS
type 8 [LQT8]), and transposition of great arteries
(n=1; genotype negative). One patient had bilateral
sensorineural hearing loss and was subsequently
confirmed to have LQTS type 1 (LQT1).
Mode of presentation
Figure 1 illustrates the mode of initial presentation
of our patients. Forty-four patients had clinically
important presentation at diagnosis. Syncope
without convulsion was the most common mode
of presentation (37.3%), among which around 40%
of cases were stress-related. Convulsion was also a
common symptom (12%). Aborted cardiac arrest
occurred in six (10.2%) individuals.
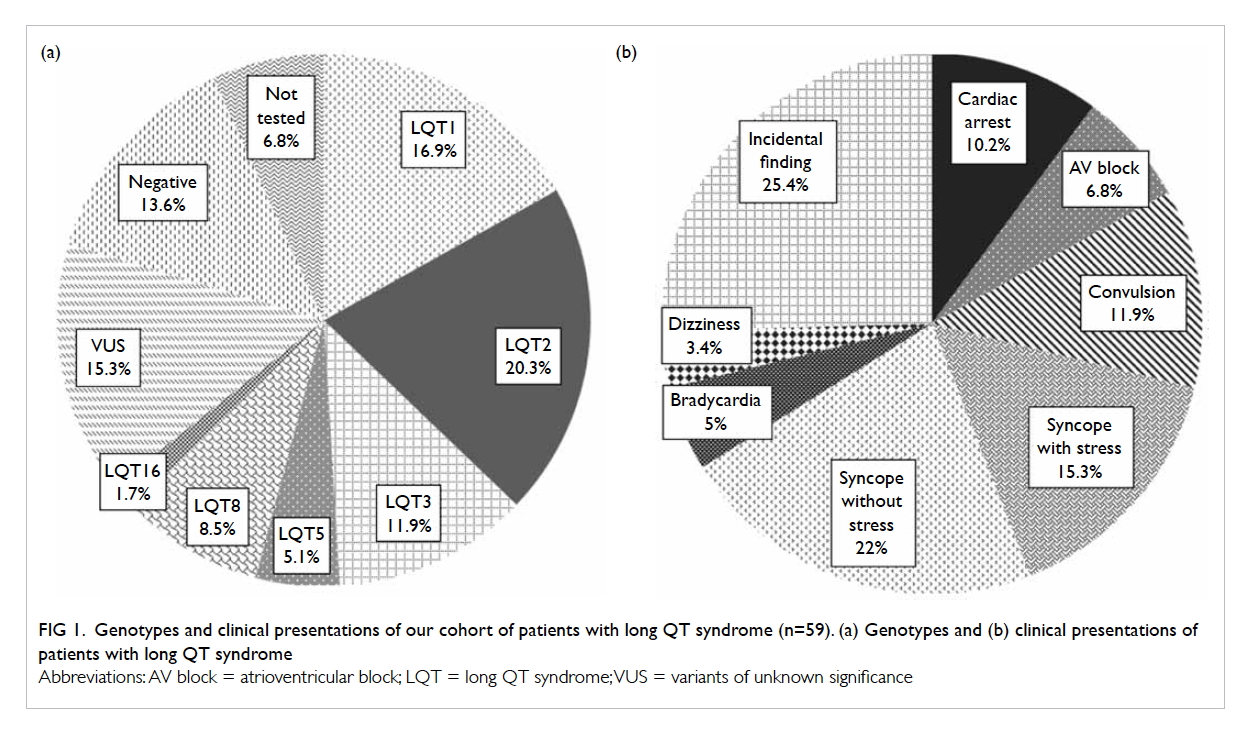
Figure 1. Genotypes and clinical presentations of our cohort of patients with long QT syndrome (n=59). (a) Genotypes and (b) clinical presentations of patients with long QT syndrome
Three patients presented with sinus
bradycardia, one of whom was detected prenatally.
Four patients, including three infants, had 2:1
atrioventricular (AV) block at diagnosis. A
significant proportion of patients with LQTS (25.4%)
did not have clinically important presentation at
diagnosis; most of them had incidental ECG findings
of prolonged QTc during medical check-ups or were
identified by family cascade screening.
Treatment and outcome
The clinical outcomes of patients with LQTS in our
cohort with clinically important presentation are
summarised in Figure 2.
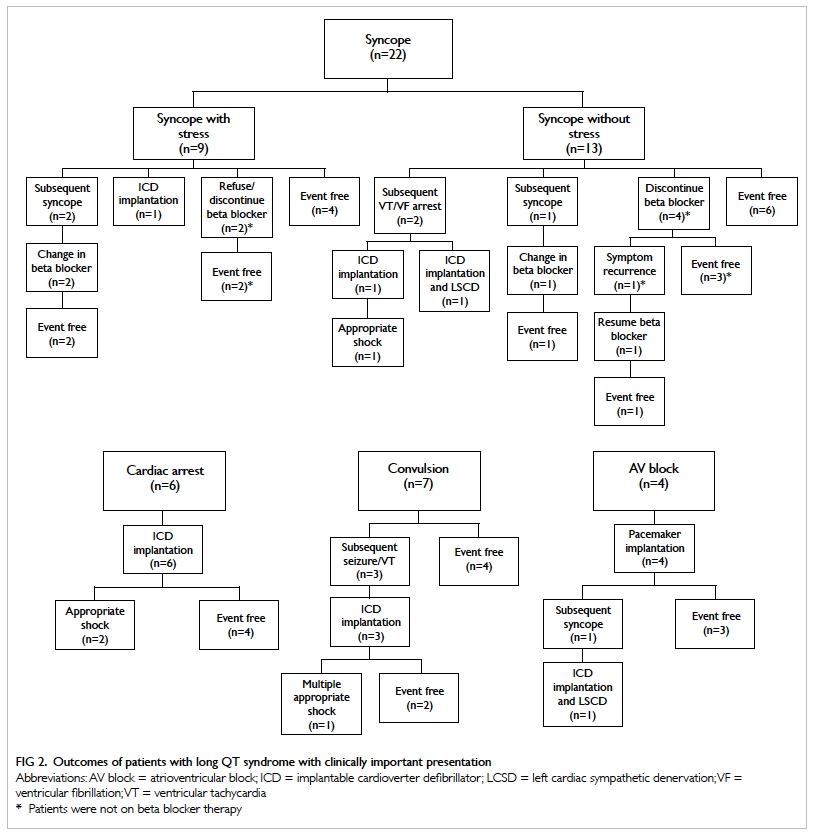
Figure 2. Outcomes of patients with long QT syndrome with clinically important presentation
Beta blocker
All patients were offered beta blocker therapy.
However, 14 patients refused beta blocker (11
patients had clinically important presentation).
Seven patients were on beta blocker but stopped
subsequently.
Metoprolol was the initial choice of beta
blocker for most of our patients (n=27), whereas 10
patients had atenolol as initial choice. Propranolol
was used in eight infants. Eight patients receiving
metoprolol, atenolol, or propranolol later changed to
nadolol to enhance compliance or for better control
of breakthrough symptoms. Mexiletine was added
as an adjuvant therapy for five patients with LQTS
type 3 (LQT3) who were symptomatic. Among the
31 patients with an initial history of convulsion,
syncope, or dizziness (mean follow-up duration,
4.81 ± 3.84 years), 21 became asymptomatic after
medication and/or lifestyle modification. Two
patients who had recurrent symptoms after initial
beta blocker therapy became event-free after
a change from metoprolol/atenolol to nadolol.
Patients without clinically important presentation at
diagnosis remained asymptomatic with or without
beta blocker therapy, irrespective of presence of
documented pathogenic mutation.
Implantable cardioverter defibrillator
Implantable cardioverter defibrillator was implanted
in 13 patients, whose mean QTc was 501 ± 42 ms. Six
of these patients had initially presented with aborted
ventricular tachycardia (VT)/ventricular fibrillation
(VF) arrest. Five patients received ICD implantation
because of recurrent symptom or subsequent VT/VF despite beta blocker therapy. Two of these five
patients experienced appropriate shocks after
ICD implantation. One patient with pathogenic
KCNE1 mutation had syncope due to sinus arrest
with long pauses; ICD was offered for primary
prevention of sudden death in addition to pacing
therapy. One patient who had AV block at birth
developed subsequent unprovoked syncope at aged
6 years despite medical treatment and his pacing
system was upgraded to ICD. In total, four patients
experienced appropriate ICD shocks despite beta
blocker treatment. There were no more ICD shocks
after reinforcement of medication compliance,
adjustment of dosage, and in one patient switching
of metoprolol to nadolol.
Left cardiac sympathetic denervation
Left cardiac sympathetic denervation was performed
via video-assisted thoracoscopic approach in two
patients, together with ICD therapy. Both of them
were free of cardiac events on follow-up.
Pacemaker
Pacemakers were implanted in five patients. Four
of these five patients had functional AV block due
to prolonged QTc. Normal AV node conduction
recovered with time in these four children. The fifth
patient had complete heart block after surgical repair
of congenital heart condition (transposition of great
arteries with ventricular septal defect).
The Kaplan-Meier survival curve is shown in
Figure 3. Overall, the breakthrough cardiac event–free survival was 93.0% ± 0.034% at 1 year, 80.7% ±
0.065% at 5 years, and 72.6% ± 0.080% at 10 years
for the entire cohort. Patients who had clinically
important presentation at baseline had a higher risk
of developing breakthrough cardiac events (P=0.048)
compared with those who did not. There was no
mortality during the study period.
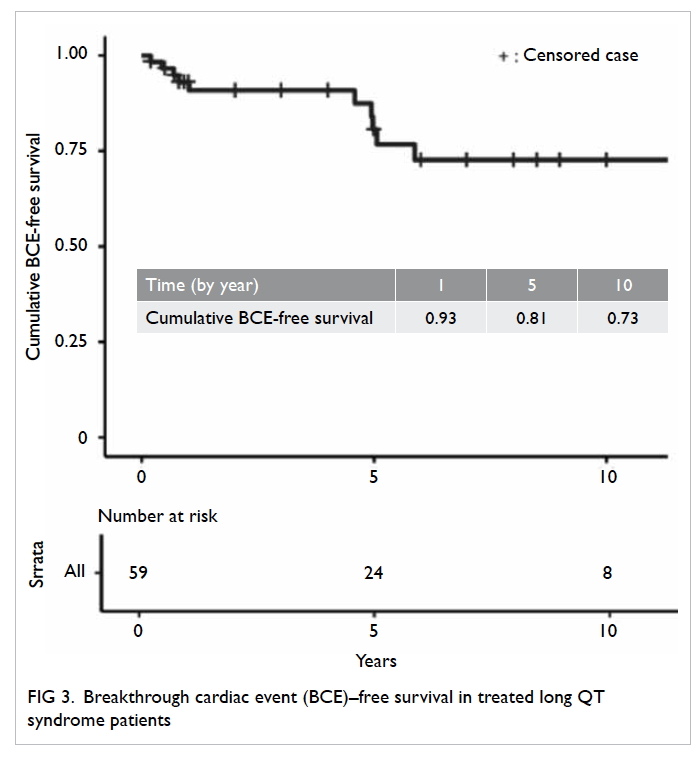
Figure 3. Breakthrough cardiac event (BCE)–free survival in treated long QT syndrome patients
Genotype
All but four patients underwent genetic testing.
Testing was not possible in two patients owing to loss
to follow-up before genetic testing could be offered;
these two patients were strong phenotypes of LQTS
with Schwartz scores of 4 and 5 points, respectively.
The other two patients were first-degree relatives of
confirmed genotype-positive patients.
Seven patients were genotype negative. Four
of them were tested before 2014. Nine patients had
their mutated genes classified as variants of unknown
significance. We also included three patients (LQT1,
n=2; LQT2, n=1), reported by Mak et al,7 whose
genetic tests were performed in a local laboratory.
Thirty-eight (69.1%) patients among those
tested were confirmed to have pathogenic mutations
for LQTS (Table 2). Eight mutations were novel
(8/33, 24.2%). Most of the pathogenic mutations
were missense mutation (30/33, 90.9%). Ten (16.9%)
patients had LQT1 (KCNQ1). Twelve (20.3%)
patients had LQT2 (KCNH2), whereas seven (11.9%)
patients had LQT3 (SCN5A). Three patients had
LQTS type 5 (KCNE1). There were five patients
(three families) with LQT8 resulting from a rare
pathogenic mutation in CACNA1C. LQT8 is linked
to Timothy syndrome with multiple extracardiac
manifestations. Only one of our patients with LQT8
had classical features of Timothy syndrome, with
syndactyly, developmental delay, and congenital
heart defect (tetralogy of Fallot). One patient
had LQTS type 16 (CALM3). Figure 1 shows the
details of the genotype information of LQTS genes
identified.
Clinical characteristics of patients with LQT1,
LQT2, or LQT3 are detailed in Figure 4. Syncope
with stress was the predominant presentation in
patients with LQT1 (60%). In contrast, syncope
without stress was the main form of presentation in
patients with LQT2 (41.7%). In patients with LQT3,
around 30% had an initial presentation of aborted VF
cardiac arrest. A high proportion (50%) of patients
with LQT3 developed subsequent VT/VF despite
medical therapy, compared with 14.3% in patients
with LQT1 and 28.6% in patients with LQT2.
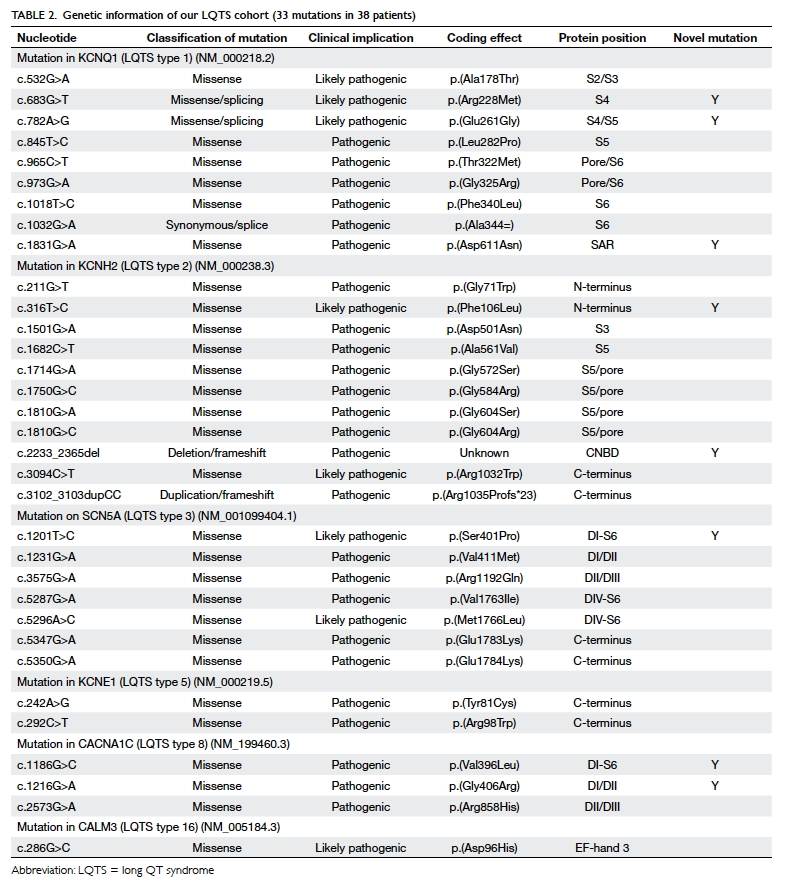
Table 2. Genetic information of our LQTS cohort (33 mutations in 38 patients)
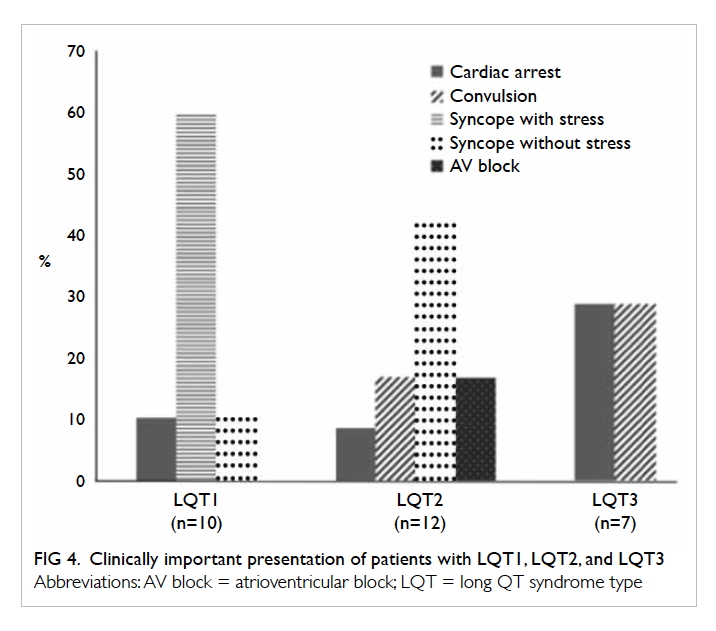
Figure 4. Clinically important presentation of patients with LQT1, LQT2, and LQT3
Nine patients were identified to have LQTS
during family screening of index patients. Two
patients had strong phenotypes but did not have
genetic tests. Five patients had LQTS phenotypes with
confirmation by genetic testing. Two asymptomatic
children in the same family were referred to us and
were identified by cascade screening of their father’s
pathogenic mutation.
Discussion
Long QT syndrome is a rare inherited disorder
associated with an increased propensity to
polymorphic VT/VF, syncope, and sudden cardiac
death. In this report, we describe the clinical features
and genetic profile of 59 LQTS paediatric patients
managed in the only tertiary paediatric cardiology
referral centre in Hong Kong over 20 years.
Long QT syndrome diagnosis
The prevalence of childhood LQTS was estimated
to be 1:2000 in an Italian birth cohort.8 In a
recent Japanese study, the estimated probability
of diagnosing LQTS was 1:3300 in children aged
6 years and 1:1000 in those aged 12 years.9 Our
results indicate that the prevalence of diagnosed
LQTS in Hong Kong children was less than 1:10 000,
suggesting an underdiagnosis of the condition. This
is likely due to under-recognition of symptomatic
LQTS in young patients who were treated for
recurrent seizure and unexplained syncope. Without
an ECG screening programme, many asymptomatic
LQTS children also remain undiagnosed.
The lack of comprehensive screening for family
members of adult LQTS probands is another reason
for underdiagnosis, as only two children from a single
family were referred to us from adult cardiologists
over the 20-year study period. In addition, a 5-year-old
child of a mother with known LQTS was not
referred until he presented with convulsions.
Molecular autopsy for young victims of
sudden cardiac death is not implemented in Hong
Kong. In many developed countries, affected
family members of LQTS sudden death victims are
identified early through this pathway to prevent
sudden cardiac death. We believe that the total
number of symptomatic and asymptomatic young
patients with LQTS in Hong Kong is much higher
than what we have studied in our single tertiary
referral centre.10 11
Long QT syndrome presentation
Congenital LQTS is usually diagnosed in patients
presenting with syncope, unexplained seizure, and
aborted cardiac arrest. The mean QTc of our patients
was 504 ± 47 ms, and median Schwartz score of the
entire cohort was 4 points (range, 1-6 points). This
indicates that the patients that were referred to our
centre were patients with more severe symptoms,
resulting in a higher likelihood of a diagnosis of
LQTS, based on clinical criteria.
Previous reports have shown that the risk of
clinical events in boys (aged <15 years) with LQTS
is significantly higher than that in girls with LQTS.12
In our cohort, we also confirmed that symptomatic
boys were significantly younger than girl at diagnosis
or presentation.
Sinus bradycardia and functional AV block are
well reported in perinatal LQTS.13 14 15 The youngest
patient in our group presented with fetal bradycardia.
We also noted 2:1 AV block in three infants at
presentation. Their AV conduction normalised
with a significant decrease in QTc during follow-up.
Paediatricians or family doctors should suspect
LQTS in young infants with slow heart rates.
Long QT syndrome and structural congenital
heart disease
Few links between LQTS and congenital heart disease
have been reported, apart from Timothy syndrome.
A recent single-centre review of 49 LQTS genotype-positive
patients identified 11 (22%) cases with
concomitant conotruncal anomalies and/or aortic
arch anomalies.16 In our cohort, five (8.5%) patients
with LQTS had concomitant congenital heart disease.
Two cases were diagnosed in the perioperative
period. Prolonged QTc in the context of congenital
heart disease can be confounded by several factors,
including postoperative electromechanical factors,
intrinsic, or postoperative QRS abnormalities.
Therefore, the diagnosis of LQTS could be masked
in patients with congenital heart disease. We suggest
that ECG of patients with congenital heart disease
should be evaluated carefully for QTc.
Long QT syndrome genotype
Throughout the world, 75% to 80% of patients
with LQTS have identifiable genetic mutations,
with LQT1, LQT2, or LQT3 accounting for 90% of
cases. Pathogenic LQTS genetic mutations were
identified in 69.1% of the patients in our cohort who
were tested, which is comparable with other LQTS
cohorts.17 Similarly, we had predominant genotypes
of LQT1 (10/59, 16.9%), LQT2 (12/59, 20.3%),
and LQT3 (7/59, 11.9%). In a recent single-centre
study of LQTS in China, LQT2 was also the most
common genotype.18 We also identified five (8.5%)
patients with rare CACNA1C (LQT8) mutations, all
of whom were Hong Kong Chinese. Without a study
of a large Chinese population in the past 5 years for
cross reference, we cannot be certain whether LQT8
is more prevalent in Chinese than in other ethnic
groups.
Long QT syndrome management and outcome
Beta blockers are the mainstay of treatment for all
LQTS genotypes. In a registry of 1530 patients with
LQTS, all beta blockers seemed equally effective in
reducing risk of a first cardiac event after beta blocker
initiation. For patients with LQT1, no single type
of beta blocker has been found superior, although
nadolol was found to be superior for patients
with LQT2.19 However, another study suggested
that symptomatic LQT1 and LQT2 patients on
metoprolol had a higher rate of recurrence of
cardiac events.20 In the present study, 21 patients
were prescribed metoprolol, two of whom required
switching to another beta blocker due to recurrent
symptoms. Because of the relatively short follow-up
duration and small number of patients in
our cohort, it is impossible to conclude on the
efficacy of each beta blocker for patients with each
genotype. Genotype-guided therapy is advocated in
contemporary management in LQTS. Based on the
available evidence, we are inclined to use nadolol
as our first choice in symptomatic LQT2 patients.
In symptomatic LQT3 patients, dual therapy using
beta blockers and mexiletine are used. Mexiletine is
a sodium channel blocker shown to shorten QTc in
LQT3 patients.21 22
In addition to medical therapy with beta
blockers, treatment of LQTS can also include lifestyle
modifications, sympathetic denervation, and device
therapy. With a multi-modality management strategy
and genotype-guided therapy, outcome of LQTS
have improved markedly over the past decades.
The event-free survival was 96% at 1 year, 93% at
5 years, and 90% at 10 years, as reported recently
in a large single-centre study which included 83%
asymptomatic probands.23 In the present study, the
breakthrough cardiac event-free survival was 93.0%
at 1 year, 80.7% at 5 years, and 72.6% at 10 years (1). The event rate in the present study is likely higher
than that of the abovementioned study because
we have a higher proportion of probands (85%),
of whom 88% were symptomatic at presentation.
Breakthrough cardiac events were mainly related to
non-compliance to our treatment advice.
Study limitations
Our study was based on single-hospital data and
referral bias is expected. We may have received
referral of patients with LQTS with more severe
symptoms. In addition, a short duration of follow-up
in our patients (mean 5.3 years) may have led
to underestimation of clinical cardiac events and
mortality.
Future perspectives
Our study demonstrated the high yield of genetic
testing and the importance of genetic information
in predicting the prognosis of patients with LQTS
and guiding their treatment. Early identification of
affected family members through cascade screening
of mutated gene was also demonstrated. We hope
that public genetic services can continue to develop,
to enable genetic testing to be offered to all patients
with suspected channelopathies. We also advocate
the establishment in Hong Kong of inherited
arrhythmia clinics or cardiac genetic clinics in
the public sector, as implemented in many other
countries. Such clinics have proven effectiveness
in reducing sudden cardiac death associated with
inherited arrhythmia syndrome.24
Conclusion
Our study provides insight into the clinical and
molecular profiles of young patients with LQTS in the
only tertiary paediatric cardiology referral centre in
Hong Kong. The LQT1, LQT2 and LQT3 genotypes
are the most common in mutation-positive patients.
Early identification of LQTS, administration of
beta blocker therapy, device therapy, and lifestyle
modifications can prevent sudden cardiac death.
However, the breakthrough cardiac event survival
was only 72.6% at 10 years. Further optimisation of
the treatment strategy by genotype-guided therapy
may reduce recurrent symptoms and improve
prognosis.
Author contributions
Concept and design: APY Liu, BHY Chung, TC Yung.
Acquisition of data: SY Kwok, CYY Chan, TC Yung.
Analysis or interpretation of data: SY Kwok, CYY Chan, JLF Fung, CCY Mak.
Drafting of the article: SY Kwok, APY Liu.
Critical revision for important intellectual content: BHY Chung, KS Lun, TC Yung.
Acquisition of data: SY Kwok, CYY Chan, TC Yung.
Analysis or interpretation of data: SY Kwok, CYY Chan, JLF Fung, CCY Mak.
Drafting of the article: SY Kwok, APY Liu.
Critical revision for important intellectual content: BHY Chung, KS Lun, TC Yung.
Acknowledgement
We would like to express our gratitude to all paediatricians
and physicians who referred patients with LQTS to our
department, and to the adult cardiologists who cared for our
grown-up patients. We are also grateful for the contributions
of the local university laboratories and the pathology
departments of Hospital Authority hospitals in conducting
genetic tests on some of our patients. We would like to express
our gratitude to Ms Yo-yo WY Chu for her participation in
providing genetic services to our patients.
Declaration
All authors have disclosed no conflicts of interest. All authors
had full access to the data, contributed to the study, approved
the final version for publication, and take responsibility for its
accuracy and integrity.
Results of this study were presented in the following
meetings: (1) World Congress of Pediatric Cardiology &
Cardiac Surgery 2017, Barcelona, Spain, 16-21 Jul 2017; (2)
The 13th Congress of Asian Society for Pediatric Research
2017, Hong Kong, 6-8 Oct 2017; (3) The 26th Annual Scientific
Congress, Hong Kong College of Cardiology 2018, Hong
Kong, 15-17 Jun 2018. Abstract of this study was published in
the Journal of the Hong Kong College of Cardiology (Kwok SY,
Liu AP, Lun KS, et al. Clinical and genetic profile of congenital
long QT syndrome in Hong Kong—18-year experience in
pediatrics. J HK Coll Cardiol 2018;26:60).
Support/funding
The expenses of the genetic analysis used in our study were
sponsored by the Children’s Heart Foundation of Hong Kong.
Ethical approval
This study received ethics approval from the Institutional
Review Board of the University of Hong Kong/Hospital
Authority Hong Kong Western Cluster.
References
1. Hobbs JB, Peterson DR, Moss AJ, et al. Risk of aborted
cardiac arrest or sudden cardiac death during adolescence
in the long-QT syndrome. JAMA 2006;296:1249-54. Crossref
2. Weintraub RG, Gow RM, Wilkinson JL. The congenital
long QT syndromes in childhood. J Am Coll Cardiol
1990;16:674-80. Crossref
3. Mizusawa Y, Horie M, Wilde A. Genetic and clinical
advances in congenital long qt syndrome. Circ J
2014;78:2827-33. Crossref
4. Giudicessi JR, Ackerman MJ. Calcium revisited: new
insights into the molecular basis of long-QT syndrome.
Circ Arrhythm Electrophysiol 2016;9:e002480. Crossref
5. Priori SG, Blomström-Lundqvist, Mazzanti A, et al. 2015
ESC Guidelines for the management of patients with
ventricular arrhythmias and the prevention of sudden
cardiac death: The Task Force for the Management of
Patients with Ventricular Arrhythmias and the Prevention
of Sudden Cardiac Death of the European Society of
Cardiology (ESC). Endorsed by: Association for European
Paediatric and Congenital Cardiology (AEPC). Eur Heart J
2015;36:2793-867. Crossref
6. Schwartz PJ, Crotti L. QTc behavior during exercise and
genetic testing for the long-QT syndrome. Circulation
2011;124:2181-4. Crossref
7. Mak CM, Chen SP, Mok NS, et al. Genetic basis of
channelopathies and cardiomyopathies in Hong Kong
Chinese patients: a 10-year regional laboratory experience.
Hong Kong Med J 2018;24:340-9. Crossref
8. Schwartz PJ, Stramba-Badiale M, Crotti L, et al. Prevalence
of the congenital long-QT syndrome. Circulation
2009;120:1761-7. Crossref
9. Yoshinaga M, Kucho Y, Nishibatake M, Ogata H, Nomura
Y. Probability of diagnosing long QT syndrome in children
and adolescents according to the criteria of the HRS/EHRA/APHRS expert consensus statement. Eur Heart J
2016;37:2490-7. Crossref
10. Kwok SY, Pflaumer A, Pantaleo SJ, Date E, Jadhav M, Davis
AM. Ten-year experience in atenolol use and exercise
evaluation in children with genetically proven long QT
syndrome. J Arrhythmia 2017;33:624-9. Crossref
11. Marcondes L, Crawford J, Earle N, et al. Long QT molecular
autopsy in sudden unexplained death in the young (1-40
years old): lessons learnt from an eight year experience in
New Zealand. PLoS One 2018;13:e0196078. Crossref
12. Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll
Cardiol 2008;51:2291-300. Crossref
13. Cuneo BF, Strasburger JF, Yu S, et al. In utero diagnosis of
long QT syndrome by magnetocardiography. Circulation
2013;128:2183-91. Crossref
14. Mitchell JL, Cuneo BF, Etheridge SP, Horigome H, Weng
HY, Benson DW. Fetal heart rate predictors of long QT
syndrome. Circulation 2012;126:2688-95. Crossref
15. Horigome H, Nagashima M, Sumitomo N, et al. Clinical
characteristics and genetic background of congenital
long-QT syndrome diagnosed in fetal, neonatal, and
infantile life a nationwide questionnaire survey in Japan.
Circ Arrhythmia Electrophysiol 2010;3:10-7. Crossref
16. Ebrahim MA, Williams MR, Shepard S, Perry JC. Genotype
positive long QT syndrome in patients with coexisting
congenital heart disease. Am J Cardiol 2017;120:256-61. Crossref
17. Schwartz PJ, Priori SG, Spazzolini C, et al. Genotype-phenotype
correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias.
Circulation 2001;103:89-95. Crossref
18. Gao Y, Liu W, Li C, et al. Common genotypes of long
QT syndrome in China and the role of ECG prediction.
Cardiology 2016;133:73-8. Crossref
19. Abu-Zeitone A, Peterson DR, Polonsky B, McNitt S, Moss
AJ. Efficacy of different beta-blockers in the treatment of
long QT syndrome. J Am Coll Cardiol 2014;64:1352-8. Crossref
20. Chockalingam P, Crotti L, Girardengo G, et al. Not all
beta-blockers are equal in the management of long QT
syndrome types 1 and 2 higher recurrence of events under
metoprolol. J Am Coll Cardiol 2012;60:2092-9. Crossref
21. Wilde AA, Moss AJ, Kaufman ES, et al. Clinical aspects of
type 3 long-QT syndrome: an international multicenter
study. Circulation 2016;134:872-82. Crossref
22. Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating
properties of SCN5A mutations and the response
to mexiletine in long-QT syndrome type 3 patients.
Circulation 2007;116:1137-44. Crossref
23. Rohatgi RK, Sugrue A, Bos JM, et al. Contemporary
outcomes in patients with long QT syndrome. J Am Coll
Cardiol 2017;70:453-62. Crossref
24. Adler A, Sadek MM, Chan AY, et al. Patient outcomes from
a specialized inherited arrhythmia clinic. Circ Arrhythm
Electrophysiol 2016;9:e003440. Crossref