DOI: 10.12809/hkmj187215
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
MEDICAL PRACTICE CME
Guidance on the management of familial
hypercholesterolaemia in Hong Kong: an expert panel consensus viewpoint
Brian Tomlinson, MB, BS, MD1; Juliana
CN Chan, MB, ChB, MD1; WB Chan, MB, ChB, FHKAM (Medicine)2;
Walter WC Chen, MD, FACC3; Francis CC Chow, MB, BS1;
SK Li, FRCP (Lond), FACC4; Alice PS Kong, FRCP, MD1;
Ronald CW Ma, FHKCP, FHKAM (Medicine)1; David CW Siu, MB, BS,
MD5; Kathryn CB Tan, MBBCH, MD5; Lawrence KS Wong,
FRCP (Lond), MD1; Vincent TF Yeung, FHKAM (Medicine), MD6;
Betty WM But, MB, BS, FHKAM (Paediatrics)7; PT Cheung, FRCP
(Edin), FHKCPaed8; CC Fu, MB, ChB, FHKAM (Paediatrics)9;
Joanna YL Tung, MB, BS, FHKAM (Paediatrics)10; WC Wong, FHKAM
(Paediatrics), FHKCPaed11; HC Yau, FHKCPaed, FHKAM
(Paediatrics)12
1 Department of Medicine and
Therapeutics, The Chinese University of Hong Kong, Shatin, Hong Kong
2 Qualigenics Diabetes Centre, Hong Kong
3 The Heart Clinic, Hong Kong
4 Premier Medical Center, Hong Kong
5 Department of Medicine, The University
of Hong Kong, Pokfulam, Hong Kong
6 Department of Medicine and Geriatrics,
Our Lady of Maryknoll Hospital, Wong Tai Sin, Hong Kong
7 Department of Paediatrics, Queen
Elizabeth Hospital, Jordan, Hong Kong
8 Town Health International Health
Management Centre, Hong Kong
9 Department of Paediatrics and
Adolescent Medicine, Princess Margaret Hospital, Laichikok, Hong Kong
10 Department of Paediatrics and
Adolescent Medicine, The University of Hong Kong, Pokfulam, Hong Kong
11 Department of Paediatrics and
Adolescent Medicine, Alice Ho Miu Ling Nethersole Hospital, Tai Po, Hong
Kong
12 Department of Paediatrics, Prince of
Wales Hospital, Shatin, Hong Kong
Corresponding author: Prof Brian Tomlinson (btomlinson@cuhk.edu.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
In 2016, meetings of groups of physicians and
paediatricians with a special interest in lipid disorders and familial
hypercholesterolaemia were held to discuss several domains of management
of familial hypercholesterolaemia in adults and children in Hong Kong.
After reviewing the evidence and guidelines for the diagnosis,
screening, and management of familial hypercholesterolaemia, consensus
was reached on the following aspects: clinical features, diagnostic
criteria, screening in adults, screening in children, management in
relation to target plasma low-density lipoprotein cholesterol levels,
detection of atherosclerosis, lifestyle and behaviour modification, and
pharmacotherapy.
Introduction
Familial hypercholesterolaemia (FH), an autosomal
codominant inherited disorder of lipoprotein metabolism, is characterised
by markedly elevated plasma low-density lipoprotein cholesterol (LDL-C)
levels and increased risk of premature atherosclerotic cardiovascular
disease (CVD), particularly coronary heart disease (CHD).1 2 3 Familial hypercholesterolaemia is generally caused by
mutations in the genes related to the LDL receptor (LDLR) pathway
(eg, loss-of-function mutations in the LDLR or apolipoprotein B
(apoB) gene (APOB) or gain-of-function mutations in the proprotein
convertase subtilisin-kexin type 9 [PCSK9] gene) resulting in
marked elevation of plasma LDL-C levels from birth.
Heterozygous (He) FH is one of the most common
human genetic disorders. It affects 1 in 200 to 300 individuals in
unselected general populations. The prevalence of homozygous (Ho) FH has
been estimated at 1 in 1 000 000, based on a frequency of 1 in 500 for
HeFH, but it is likely to be more common.1
4 Familial hypercholesterolaemia is
associated with considerable morbidity and mortality because of CHD. If
left untreated, men and women with HeFH typically develop CHD before the
ages of 55 and 60 years, respectively; 50% of men and 15% of women die
before these ages, whereas those with HoFH may develop CHD very early in
life.1
Early identification and optimal treatment of
patients with FH are crucial for the prevention of atherosclerosis
progression and coronary complications. Although FH is a very common
genetic disorder, it remains largely undetected and undertreated.1 Recent guidelines and consensus statements in Europe
and in some Asia-Pacific countries highlight the need for the early
identification of FH to improve the awareness and management of this
condition.1 4 5 6 7
As in most other countries,8 there are significant gaps in the awareness and
management of FH among physicians and the general public in Hong Kong.
There is no clear management guideline or consensus statement for FH in
Hong Kong. On 22 February 2016, 12 experts on lipid disorders in Hong Kong
convened for the local Advisory Board Meeting on the Management of
Familial Hypercholesterolaemia in Hong Kong; and on 14 December 2016, 10
experts convened for a second meeting specifically regarding the
management of paediatric patients with FH in Hong Kong. The principal
objectives were to discuss the evidence for diagnosis, screening, and
management of FH, in order to develop a consensus statement relevant to
Hong Kong. The panel reviewed both international guidelines and those for
individual Asia-Pacific countries, then developed a consensus treatment
matrix/guide regarding the diagnosis, screening, and management of FH. The
expert panel discussed each issue until they had attained a unanimous
consensus.
Clinical features of familial hypercholesterolaemia in
Hong Kong
Plasma LDL-C levels in the Hong Kong general
population are comparable to those of some Western countries.9 10 According
to the experts’ clinical experience, patients with FH in Hong Kong,
especially older adults, tend to exhibit CVD later in life (approximately
70 years of age), compared with patients in Western countries. Many older
patients with FH in Hong Kong are free of cardiovascular events in their
70s or 80s; this may be related to their previously healthy lifestyle (eg,
substantial physical activity with a healthy diet). However, young
patients with FH tend to develop CVD at an earlier age than older patients
within the same families. More recently, cardiovascular events have been
observed in patients who are in their mid-20s. The increased risk in these
young patients is likely due to lifestyle changes in the younger
generations. Stroke remains uncommon in patients with FH in Hong Kong,
presumably because elevated LDL-C levels are not a strong risk factor for
cerebrovascular diseases.11
Clinical characteristics have been reported for 252
Hong Kong Chinese patients from 87 pedigrees who were clinically diagnosed
with FH during 1990-2000 (mean [standard deviation] age 37 [17] years,
including 43 patients aged <18 years).10
The mean plasma LDL-C level was 7.2 (1.5) mmol/L.10
Tendon xanthomata was present in 40.6% of males and 54.8% of females. The
prevalence of known CHD was relatively low: 9.9% in males and 8.5% in
females.10
Diagnostic criteria of familial hypercholesterolaemia
Although FH is generally considered to be a
monogenic condition, it is typically diagnosed on the basis of clinical
features and family history, rather than a genetic test. There are several
sets of clinical criteria for diagnosing FH (Table 17 12 13
14 15),
including the Simon Broome Register diagnostic criteria,12 the Make Early Diagnosis to Prevent Early Deaths
(MEDPED) criteria,13 and the Dutch
Lipid Clinic Network Diagnostic Criteria14
(DLCNC; online supplementary Appendix); however, none of these are
universally accepted as the best approach. More recently, Japanese experts
have developed specific criteria for the diagnosis of FH in Japan.7
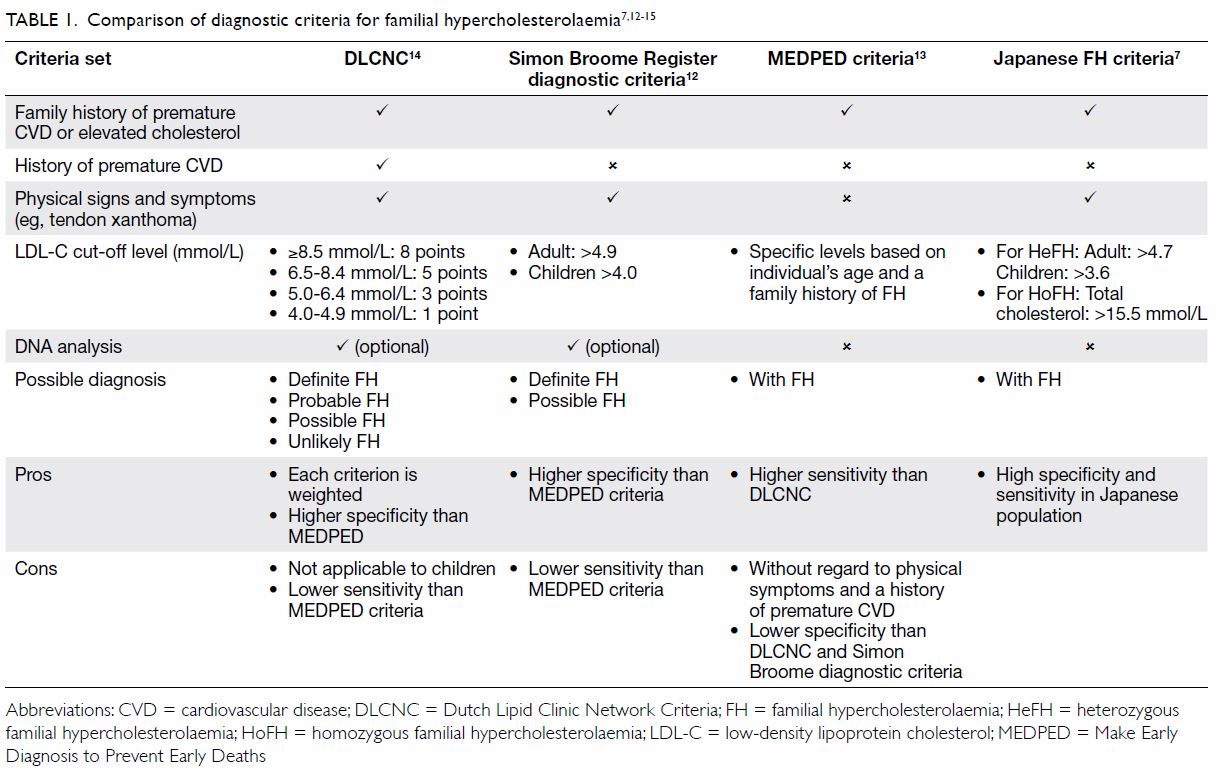
The DLCNC14
use a point system to assess the following characteristics: family history
of FH, history of premature CVD, physical examination of tendinous
xanthomata and premature arcus cornealis, LDL-C levels, and DNA analysis.
There is a point score for each item; a total point score of >8 is
regarded as definite FH, 6 to 8 as probable FH, 3 to 5 as possible FH, and
<3 as unlikely FH. Similar to the DLCNC, the Simon Broome criteria12 use family history of FH, physical signs (excluding
arcus cornealis), LDL-C levels, and genetic tests to predict the
probability of the diagnosis of FH. The MEDPED criteria13 rely on plasma total cholesterol and LDL-C levels in
the probands and their family members, without consideration of other
phenotypes. The MEDPED criteria have a higher sensitivity, but lower
specificity than the Dutch and Simon Broome diagnostic criteria. The
Japanese FH criteria,16 which are
similar to the Simon Broome criteria, use a population-specific LDL-C
level >4.7 mmol/L for adults and >3.6 mmol/L for children as a
criterion for the diagnosis of FH.
The Dutch criteria were developed from patients who
had been genotyped; thus, these comprise the only set of criteria
validated by genetic tests. The panel agreed to apply the Dutch criteria
for the diagnosis of FH adults in Hong Kong; however, because of lower
reported LDL-C levels in local patients with FH, the panel recommended a
lower threshold for LDL-C levels indicative of definite FH, probable FH,
possible FH, and unlikely FH.10
Secondary causes of increased LDL-C levels, such as hypothyroidism and
nephrotic syndrome, should be excluded before considering a diagnosis of
FH.
Screening for familial hypercholesterolaemia in adults
in Hong Kong
Universal screening for FH in adults is not
practicable in Hong Kong or in most other countries. General screening for
FH as primary prevention in Hong Kong can be challenging, as it is
difficult to convince asymptomatic patients to participate in the
screening programme. A regular body check, including measurement of the
plasma lipid profile, is becoming more popular in Hong Kong. The panel
recommended that greater attention should be given to the cholesterol
profile as a routine body check item, together with documentation of
family history of FH and premature CHD; this approach may increase the
likelihood of identifying potential index FH cases. The risk of
cardiovascular events in patients with FH largely depends on the plasma
LDL-C level; however, other risk factors, such as smoking, hypertension,
diabetes, and elevated levels of lipoprotein(a) [Lp(a)], are also
important. Targeted LDL-C screening in high-risk patients, especially
younger patients with premature CHD, is encouraged.
The panel recommended that adults with a plasma
LDL-C level >5 mmol/L should be regarded as potential probands. For
patients at high risk of FH, such as patients with a family history of FH
or premature CHD, the LDL-C level threshold could be 4.5 mmol/L. Tendon
xanthomata, arcus cornealis, and tuberous xanthoma or xanthelasma are
typically observed in patients with FH who exhibit very high LDL-C levels.
Xanthelasma and arcus cornealis are not specific clinical signs for FH.
Tendon xanthomas are more specific for FH and occur in patients with
markedly elevated LDL-C levels (typically >7.0 mmol/L); these are
rarely present before adulthood in patients with HeFH. They can also occur
in patients with sitosterolaemia and cerebrotendinous xanthomatosis.
Cascade screening for relatives of patients with FH
is recommended in both the private and public sectors. Although this may
be challenging in the private sector due to financial constraints, cascade
screening is the most cost-effective approach for the identification of
new patients with FH; moreover, it is recommended by international and
national bodies, such as the European Atherosclerosis Society and the
American Heart Association.1 5 The relatives of patients with FH can be screened with
a combination of plasma lipid profiles and genetic testing. If the
causative mutation is unknown or genetic testing is unavailable, screening
can be performed by using plasma lipid profiles alone. Currently, a
potential patient with FH must wait several months for counselling and
genetic testing in the public sector (ie, the Hong Kong Department of
Health Clinical Genetic Service) and the cost of genetic testing may not
be covered by the public health care system.
Genetic testing may not always be necessary or
cost-effective. Patients with high LDL-C levels typically must be treated,
regardless of the genetic test results; notably, these test results may
not substantially alter treatment strategies. Although there may not be
great advantages to genetic testing, there are potential benefits in
genotyping.17 For similar LDL-C
levels, the risk of cardiovascular events is greater in patients with FH
than in those without, due to their lifelong exposure to high LDL-C levels
since birth. Treatment may not be necessary in patients with FH who have
mildly elevated LDL-C levels. In contrast, long-term follow-up is
necessary in patients with FH who have similar LDL-C levels. With the
increasing affordability of genetic testing, the resulting genetic
information will help improve the precision of diagnosis and management of
FH.
Screening for familial hypercholesterolaemia in
children in Hong Kong
Universal screening of plasma cholesterol levels in
children has been proposed in some Western countries, including Australia18 and the US.19 20 Early
diagnosis can lead to effective treatment with lifestyle modification and
pharmacotherapy, as appropriate. By reducing the lifetime exposure to
LDL-C from an early age, these patients experience substantial benefits in
terms of CVD prevention. Thus, universal cholesterol screening in children
is more cost-effective than identical screening in younger or older
adults. Although it is expensive, universal cholesterol screening in
childhood may offer the best and most effective strategy for diagnosing
FH.18 The paediatric panel agreed
that universal screening should target all citizens below 20 years of age,
ideally before puberty; moreover, it should identify potential cases of FH
based on age- and gender-specific plasma LDL-C levels.
Cascade screening is highly recommended in children
with elevated LDL-C levels and in children with relatives who exhibit FH
phenotypes. Children with a relevant family history and an LDL-C level
>3.6 mmol/L are likely to have FH. In a local survey of Chinese
adolescents in Hong Kong (median [interquartile range] age, 16 [14-17]
years), the mean (standard deviation) LDL-C level was 2.15 (0.60) mmol/L
in boys and 2.24 (0.61) mmol/L in girls; thus, the 95th percentile would
be approximately 3.4 mmol/L.21 In
children with a plasma LDL-C level >4.9 mmol/L and/or physical signs
(eg, xanthomata), FH is likely; these children should be screened at any
age, as soon as they are identified. Because FH and sitosterolaemia share
several clinical characteristics, sitosterolaemia should also be
considered in these patients, especially if both parents appear to exhibit
normal lipid levels. Sitosterolaemia can be identified by measuring the
plasma levels of plant sterols; the genetic defect can be detected by
sequencing the genes for the ABCG5 and ABCG8 transporters.22 After consideration of international recommendations
and the increasingly early age of acquisition of other risk factors,
including obesity and diabetes, in our local population, the paediatric
panel suggested a screening age of 5 to 10 years to identify FH; moreover,
the panel suggested that a lower threshold for LDL-C levels should be used
in children, relative to that used in adults. The paediatric panel also
agreed that genetic testing, if available, should be provided for all
children who are suspected to have FH, after counselling. Genetic testing
would be particularly useful in children whose LDL-C levels are not
sufficiently high to make a definite diagnosis of FH when a mutation has
been detected in an affected parent or sibling. Genetic counselling should
be provided to the family before undergoing genetic testing to ensure a
clear understanding of the implications of such tests.
Despite these recommendations, the panel emphasised
that additional surveys are required regarding the distribution of plasma
cholesterol levels among local children, in order to improve the screening
strategy for FH in children.
Management of familial hypercholesterolaemia
Target plasma low-density lipoprotein cholesterol
levels
The prognosis of FH largely depends on the plasma
LDL-C levels; these should be maintained as low as possible. The panel
suggested that, for primary prevention of CHD, the target LDL-C level for
Hong Kong Chinese patients with FH should be <2.5 mmol/L. The panel
agreed that patients with established atherosclerotic CVD or other
cardiovascular risk factors, such as diabetes, elevated Lp(a) level ≥50
mg/dL, pretreatment LDL-C level ≥6.72 mmol/L, family history of premature
CHD, or advanced age, should be considered as very high risk. For
very-high-risk patients, the target LDL-C level should be <1.8 mmol/L;
for paediatric patients (>10 years of age) with FH, the panel
recommended that the target LDL-C level should be <3.4 mmol/L.
Detection of atherosclerosis
The detection of atherosclerosis should begin by
taking a complete medical history and performing a thorough physical
examination. If the patient is suspected to have atherosclerotic CVD, it
may be appropriate to refer them to a cardiologist or other appropriate
specialist for further investigation. Computed tomographic coronary
angiography is a useful and non-invasive tool to detect coronary
atherosclerosis and determine CVD risk. Stress echocardiography can be
used to assess myocardial functional capacity and the possibility of
silent ischaemia. Carotid ultrasound imaging is non-invasive and can
identify early-stage atherosclerosis; it can be used to assess carotid
artery disease, predict the risk of stroke, and determine the requirement
for intensive treatment in patients with FH. The ankle-brachial index is a
useful diagnostic test for early peripheral arterial disease and has been
shown to predict CVD and all-cause death in Chinese populations.23 24 Pulse
wave velocity is a non-invasive measure of arterial stiffness which also
correlates with cardiovascular events, such as the development of CHD.
Lifestyle and behaviour modification
All patients with a clinical diagnosis of FH should
be counselled on lifestyle modification, particularly healthy eating,
regular exercise and physical activity, weight control, and cessation of
smoking.
Pharmacotherapy
First-line treatment for hypercholesterolaemia for
reducing the risk of CHD involves the use of 3-hydroxy-3-methylglutaryl
coenzyme A reductase inhibitors or statins, which significantly reduce the
risk of CVD and progression of atherosclerosis in FH. In a long-term
cohort study involving more than 2000 patients with FH without prevalent
CHD in the Netherlands, patients treated with statins showed a 76% risk
reduction (hazard ratio=0.24; 95% confidence interval=0.18-0.30;
P<0.001) for CHD compared with untreated patients.25 In patients with HoFH, statin-treated patients showed
a 66% reduction in all-cause mortality and 51% reduction in major
cardiovascular events compared with statin-naïve patients; however, the
mean reduction in LDL-C level was only 26.4% with lipid-lowering therapy.26
A recent Mendelian randomisation analysis revealed
that prolonged exposure to lower LDL-C levels, beginning early in life,
reduced the risk of CHD by three-fold, when compared with the risk
reduction achieved by lowering LDL-C level with a statin started later in
life.27 The European
Atherosclerosis Society Consensus Panel recommended early detection (from
age 5 years, or earlier if HoFH is suspected) in children; the panel
suggested lifestyle modification and statin therapy for the treatment of
children with FH, as early as age 8 to 10 years.6
Typically, adult patients with FH should be treated
with high-intensity statin therapy. Female patients should be advised that
statins are contra-indicated during pregnancy and should be avoided during
lactation.5 If the target LDL-C
level cannot be achieved with statin monotherapy, a combination therapy
with concurrent ezetimibe and/or a bile-acid sequestrant or niacin can be
considered. Generally, Lp(a) levels are increased in patients with FH,28 and are considered an independent predictor of CHD in
FH after adjustment for other modifiable risk factors.1 29 30 It is desirable to measure the Lp(a) level if the
assay is available. Niacin can reduce plasma Lp(a) levels by 30% to 40%;
notably, the LDL-C level lowering-effect of niacin is largely dependent on
baseline LDL-C levels.31 32 Therefore, if available, niacin may be used in
patients with FH who do not reach their target LDL-C levels with statin
therapy. Lipoprotein apheresis will also reduce Lp(a) level, but is not
readily available in the public hospitals in Hong Kong; however,
plasmapheresis is currently used.
In Hong Kong, statins are the main therapy for
paediatric patients with FH. All available statins are approved for use in
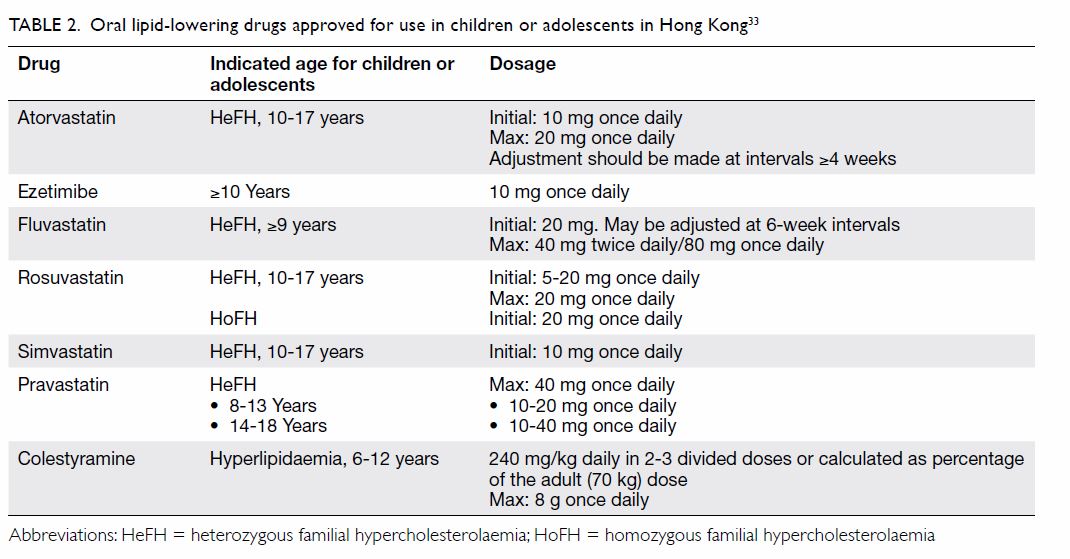
patients with HeFH aged ≥10 years (Table 233).
However, in exceptional circumstances, such as when there is a family
history of premature CHD, statins are used before age 10 years, as
recommended by the guidelines from the United Kingdom National Institute
for Health and Care Excellence.34
A 2017 Cochrane review analysed nine randomised controlled trials
comparing the efficacy and safety of statins versus placebo in 1177
children with FH aged 6 to 18 years; the authors concluded that statins
seem to be safe in the short term, but long-term safety remains unknown.35
Patients are initially treated with the lowest
doses, which can be increased as necessary. Some patients are prescribed
bile acid sequestrants (eg, colestyramine) as early as age 1 year, and
ezetimibe at age ≥10 years (Table 233).
Plasmapheresis is reserved for patients with severe disease uncontrolled
by conventional therapy. It should be emphasised that lifestyle
interventions should be the first-line treatment for paediatric patients
with FH; they should not be disregarded, even if pharmacotherapy is used.
Emerging therapies
Monoclonal antibodies to PCSK9 have emerged as the
most promising treatment option for patients with FH. This class of
agents, given by subcutaneous injection once or twice monthly, reduced
LDL-C levels by 50% to 70% in patients with HeFH who were treated with
statins with or without ezetimibe,36
37 as well as in patients with
primary hypercholesterolaemia with or without statin therapy.38 39 Two PCSK9
inhibitors, alirocumab (previously known as REGN727 and SAR236553, Sanofi
and Regeneron Pharmaceuticals, Inc) and evolocumab (AMG-145, Amgen) were
approved by the US Food and Drug Administration (FDA) and European
Medicines Agency in 2015 for their proven efficacy in reducing LDL-C
levels in patients at risk for CVD; these drugs are available in Hong
Kong. By using this group of drugs, very low LDL-C levels (eg, <1.0
mmol/L) can be achieved in patients with HeFH.
Mipomersen is an apoB antisense oligonucleotide
which inhibits the biosynthesis of apoB, thus reducing hepatic very
low–density lipoprotein cholesterol (VLDL-C) production and secretion.40 In clinical trials, subcutaneous injection of
mipomersen reduced plasma LDL-C levels by 25% and 28% in patients with
HoFH41 and HeFH,42 respectively. The major side-effects of mipomersen
include frequent injection site reactions, short-lived fatigue and
myalgia, hepatic steatosis, and elevations in plasma aminotransferases.
These hepatic changes typically resolve upon drug discontinuation.
Mipomersen is not available in Hong Kong.
Lomitapide is an orally available microsomal
triglyceride transfer protein inhibitor which decreases the hepatic
production and secretion of VLDL-C. Lomitapide has been approved for the
treatment of HoFH in the US and Europe as an add-on therapy. In a
multi-centre study of patients with HoFH, lomitapide reduced LDL-C levels
by 50%, 44%, and 38% at 26, 56, and 78 weeks, respectively.43 However, lomitapide may increase plasma
aminotransferases and intrahepatic fat content. Lomitapide is not
available in Hong Kong. Both mipomersen and lomitapide work via pathways
independent of the LDLR and are effective in patients with HoFH who
exhibit null mutations. These two drugs have been approved by the FDA for
use in patients with HoFH.
Conclusion
Patients with FH remain underdiagnosed and
undertreated in Hong Kong. Increased awareness, early identification, and
optimal treatment are essential to reduce the risk of premature CHD,
thereby restoring decades of healthy, normal life in patients with FH.
Developing a model of care for FH in Hong Kong will help to bridge the gap
in prevention of CVD and improve outcomes in patients with FH. Action is
needed to collect more population-based data to further guide
recommendations and the development of models of care for the management
of FH. While these data are gathered, this consensus statement aims to
serve as a guide to inform clinical practice and future research.
Author contributions
All authors have made substantial contributions to
the expert panel consensus viewpoint and provided critical revision for
important intellectual content. B Tomlinson is responsible for drafting of
the article.
Acknowledgement
The expert panel thanks Sanofi-Aventis Hong Kong
Limited for supporting the organisation of the meetings and providing
editorial assistance in preparing the statement by an unrestricted
educational grant.
Funding/support
The meetings during which this consensus statement
was formulated and some editorial assistance in preparing the manuscript
were funded by an unrestricted educational grant from Sanofi-Aventis Hong
Kong Limited. The funder had no role in determining the content of the
expert panel consensus statement.
Declaration
All authors have disclosed no conflicts of
interest. All authors had full access to the data, contributed to the
study, approved the final version for publication, and take responsibility
for its accuracy and integrity.
References
1. Nordestgaard BG, Chapman MJ, Humphries
SE, et al. Familial hypercholesterolaemia is underdiagnosed and
undertreated in the general population: guidance for clinicians to prevent
coronary heart disease: consensus statement of the European
Atherosclerosis Society. Eur Heart J 2013;34:3478-90a. Crossref
2. Hovingh GK, Davidson MH, Kastelein JJ,
O’Connor AM. Diagnosis and treatment of familial hypercholesterolaemia.
Eur Heart J 2013;34:962-71. Crossref
3. Reiner Ž. Management of patients with
familial hypercholesterolaemia. Nat Rev Cardiol 2015;12:565-75. Crossref
4. Cuchel M, Bruckert E, Ginsberg HN, et
al. Homozygous familial hypercholesterolaemia: new insights and guidance
for clinicians to improve detection and clinical management. A position
paper from the Consensus Panel on Familial Hypercholesterolaemia of the
European Atherosclerosis Society. Eur Heart J 2014;35:2146-57. Crossref
5. Gidding SS, Champagne MA, de Ferranti
SD, et al. The agenda for familial hypercholesterolemia: a scientific
statement from the American Heart Association. Circulation
2015;132:2167-92. Crossref
6. Wiegman A, Gidding SS, Watts GF, et al.
Familial hypercholesterolaemia in children and adolescents: gaining
decades of life by optimizing detection and treatment. Eur Heart J
2015;36:2425-37. Crossref
7. Harada-Shiba M, Arai H, Oikawa S, et al.
Guidelines for the management of familial hypercholesterolemia. J
Atheroscler Thromb 2012;19:1043-60. Crossref
8. Pang J, Sullivan DR, Harada-Shiba M, et
al. Significant gaps in awareness of familial hypercholesterolemia among
physicians in selected Asia-Pacific countries: a pilot study. J Clin
Lipidol 2015;9:42-8. Crossref
9. Pang RW, Tam S, Janus ED, et al. Plasma
lipid, lipoprotein and apolipoprotein levels in a random population sample
of 2875 Hong Kong Chinese adults and their implications (NCEP ATP-III,
2001 guidelines) on cardiovascular risk assessment. Atherosclerosis
2006;184:438-45. Crossref
10. Hu M, Lan W, Lam CW, Mak YT, Pang CP,
Tomlinson B. Heterozygous familial hypercholesterolemia in Hong Kong
Chinese. Study of 252 cases. Int J Cardiol 2013;167:762-7. Crossref
11. O’Donnell MJ, Xavier D, Liu L, et al.
Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22
countries (the INTERSTROKE study): a case-control study. Lancet
2010;376:112-23. Crossref
12. Scientific Steering Committee on
behalf of the Simon Broome Register Group. Risk of fatal coronary heart
disease in familial hypercholesterolaemia. BMJ 1991;303:893-6. Crossref
13. Williams RR, Hunt SC, Schumacher MC,
et al. Diagnosing heterozygous familial hypercholesterolemia using new
practical criteria validated by molecular genetics. Am J Cardiol
1993;72:171-6. Crossref
14. Civeira F, International Panel on
Management of Familial Hypercholesterolemia. Guidelines for the diagnosis
and management of heterozygous familial hypercholesterolemia.
Atherosclerosis 2004;173:55-68. Crossref
15. Damgaard D, Larsen ML, Nissen PH, et
al. The relationship of molecular genetic to clinical diagnosis of
familial hypercholesterolemia in a Danish population. Atherosclerosis
2005;180:155-60. Crossref
16. Harada-Shiba M, Arai H, Okamura T, et
al. Multicenter study to determine the diagnosis criteria of heterozygous
familial hypercholesterolemia in Japan. J Atheroscler Thromb
2012;19:1019-26. Crossref
17. Nordestgaard BG, Benn M. Genetic
testing for familial hypercholesterolaemia is essential in individuals
with high LDL cholesterol: who does it in the world? Eur Heart J
2017;38:1580-3. Crossref
18. Pang J, Martin AC, Mori TA, Beilin LJ,
Watts GF. Prevalence of familial hypercholesterolemia in adolescents:
potential value of universal screening? J Pediatr 2016;170:315-6. Crossref
19. Kwiterovich PO, Gidding SS. Universal
screening of cholesterol in children. Clin Cardiol 2012;35:662-4. Crossref
20. Expert panel on integrated guidelines
for cardiovascular health and risk reduction in children and adolescents:
summary report. Pediatrics 2011;128(Suppl 5):S213-56. Crossref
21. Kong AP, Choi KC, Ko GT, et al.
Associations of overweight with insulin resistance, beta-cell function and
inflammatory markers in Chinese adolescents. Pediatr Diabetes
2008;9:488-95. Crossref
22. Hu M, Yuen YP, Kwok JS, Griffith JF,
Tomlinson B. Potential effects of NPC1L1 polymorphisms in protecting
against clinical disease in a Chinese family with sitosterolaemia. J
Atheroscler Thromb 2014;21:989-95. Crossref
23. Hong Kong Diabetes Registry, Yang X,
So WY, et al. Development and validation of an all-cause mortality risk
score in type 2 diabetes. Arch Intern Med 2008;168:451-7. Crossref
24. Luo YY, Li J, Xin Y, Zheng LQ, Yu JM,
Hu DY. Risk factors of peripheral arterial disease and relationship
between low ankle brachial index and mortality from all-cause and
cardiovascular disease in Chinese patients with hypertension. J Hum
Hypertens 2007;21:461-6. Crossref
25. Versmissen J, Oosterveer DM,
Yazdanpanah M, et al. Efficacy of statins in familial
hypercholesterolaemia: a long term cohort study. BMJ 2008;337:a2423. Crossref
26. Raal FJ, Pilcher GJ, Panz VR, et al.
Reduction in mortality in subjects with homozygous familial
hypercholesterolemia associated with advances in lipid-lowering therapy.
Circulation 2011;124:2202-7. Crossref
27. Ference BA, Yoo W, Alesh I, et al.
Effect of long-term exposure to lower low-density lipoprotein cholesterol
beginning early in life on the risk of coronary heart disease: a Mendelian
randomization analysis. J Am Coll Cardiol 2012;60:2631-9. Crossref
28. Langsted A, Kamstrup PR, Benn M,
Tybjærg-Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause
of clinical familial hypercholesterolaemia: a prospective cohort study.
Lancet Diabetes Endocrinol 2016;4:577-87. Crossref
29. Alonso R, Andres E, Mata N, et al.
Lipoprotein(a) levels in familial hypercholesterolemia: an important
predictor of cardiovascular disease independent of the type of LDL
receptor mutation. J Am Coll Cardiol 2014;63:1982-9. Crossref
30. Chan DC, Pang J, Hooper AJ, et al.
Elevated lipoprotein(a), hypertension and renal insufficiency as
predictors of coronary artery disease in patients with genetically
confirmed heterozygous familial hypercholesterolemia. Int J Cardiol
2015;201:633-8. Crossref
31. Hu M, Tomlinson B. Niacin for
reduction of cardiovascular risk. N Engl J Med 2014;371:1941-2. Crossref
32. Hu M, Yang YL, Ng CF, et al. Effects
of phenotypic and genotypic factors on the lipid responses to niacin in
Chinese patients with dyslipidemia. Medicine (Baltimore) 2015;94:e881. Crossref
33. Monthly Index of Medical Specialities.
Available from: http://www.mims.com/hongkong. Accessed 1 Aug 2017.
34. National Institute for Health and
Clinical Excellence. Familial hypercholesterolaemia: identification and
management. 2008. Available from: https://www.nice.org
.uk/guidance/cg71/resources/familial-hypercholesterolaemia-identification-and-management-pdf-
975623384005.
Accessed 1 Feb 2016.
35. Vuorio A, Kuoppala J, Kovanen PT, et
al. Statins for children with familial hypercholesterolemia. Cochrane
Database Syst Rev 2017;(7):CD006401. Crossref
36. Stein EA, Gipe D, Bergeron J, et al.
Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce
low-density lipoprotein cholesterol in patients with heterozygous familial
hypercholesterolaemia on stable statin dose with or without ezetimibe
therapy: a phase 2 randomised controlled trial. Lancet 2012;380:29-36. Crossref
37. Stein EA, Honarpour N, Wasserman SM,
Xu F, Scott R, Raal FJ. Effect of the proprotein convertase
subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial
hypercholesterolemia. Circulation 2013;128:2113-20. Crossref
38. McKenney JM, Koren MJ, Kereiakes DJ,
Hanotin C, Ferrand AC, Stein EA. Safety and efficacy of a monoclonal
antibody to proprotein convertase subtilisin/kexin type 9 serine protease,
SAR236553/REGN727, in patients with primary hypercholesterolemia receiving
ongoing stable atorvastatin therapy. J Am Coll Cardiol 2012;59:2344-53. Crossref
39. Stein EA, Mellis S, Yancopoulos GD, et
al. Effect of a monoclonal antibody to PCSK9 on LDL cholesterol. N Engl J
Med 2012;366:1108-18. Crossref
40. Visser ME, Witztum JL, Stroes ES,
Kastelein JJ. Antisense oligonucleotides for the treatment of
dyslipidaemia. Eur Heart J 2012;33:1451-8. Crossref
41. Raal FJ, Santos RD, Blom DJ, et al.
Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL
cholesterol concentrations in patients with homozygous familial
hypercholesterolaemia: a randomised, double-blind, placebo-controlled
trial. Lancet 2010;375:998-1006. Crossref
42. Stein EA, Dufour R, Gagne C, et al.
Apolipoprotein B synthesis inhibition with mipomersen in heterozygous
familial hypercholesterolemia: results of a randomized, double-blind,
placebo-controlled trial to assess efficacy and safety as add-on therapy
in patients with coronary artery disease. Circulation 2012;126:2283-92. Crossref
43. Cuchel M, Blom DJ, Averna MR. Clinical
experience of lomitapide therapy in patients with homozygous familial
hypercholesterolaemia. Atheroscler Suppl 2014;15:33-45. Crossref