DOI: 10.12809/hkmj154733
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Immunoglobulin G4–related sclerosing disease involving
the mandible
Antonio CK Tong, PhD, BDS1,2; Irene OL
Ng, PhD, MD3; Miko CM Lo, MOMS RCSEd, BDS2
1 Queen Mary Hospital, Pokfulam,
Hong Kong
2 Department of Health, Hong Kong
3 Department of Pathology and State
Key Laboratory for Liver Research, The University of Hong Kong, Pokfulam,
Hong Kong
Corresponding author: Dr Antonio CK Tong (antonio.tong@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Case report
A 46-year-old female was referred by her general
dental practitioner in December 2013 for investigation of delayed healing
of lower right premolar (44, 45) socket wounds following tooth extraction
3 weeks earlier. The lower right first and second premolars—teeth 44,
45—had presented with sudden onset of pain and rapid increase in mobility
over a 2-month period. Both teeth were subsequently extracted but the
patient experienced increasing pain and discomfort around the extraction
sites.
Clinical examination showed unhealed sockets of
teeth 44, 45; with associated flabby, oedematous and verrucous gingival
tissue (Fig 1). The lower right canine tooth demonstrated
grade III mobility with radiographic evidence of periodontal bone loss.
The provisional diagnoses included eosinophilic granuloma or Langerhans
cell histiocytosis, massive osteolysis, and malignancies. Incisional
biopsy was performed under local anaesthesia. Histopathological
examination revealed infiltration by chronic inflammatory cells involving
the epithelium and stroma, with absence of malignancy.
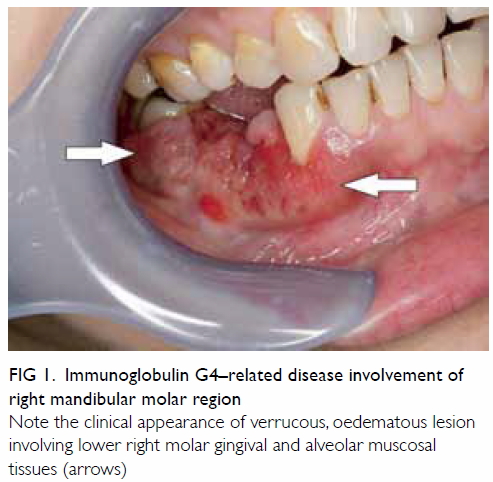
Note the clinical appearance of verrucous, oedematous lesion involving lower right molar gingival and alveolar muscosal tissues (arrows)
The symptoms persisted after the biopsy, with
progression of periodontal bone loss around the lower right canine. A
repeated biopsy was performed. Tissue was taken from the gingival
mucosa, the soft tissue around the previous extraction sockets, and the
involved mandibular bone.
Histologically, the soft tissue surrounding the
involved area comprised heavily inflamed fibrous tissue that was denuded
without epithelial covering. It contained a heavy infiltrate of
lymphocytes and plasma cells with some polymorphs. The plasma cells were
normal in appearance. The fibrous background was composed of whorls of
relatively plump fibroblasts in interlacing fascicles (Fig
2a). Frequent prominent arteries were seen but no definite
phlebitis was evident (Fig 2b). Granulomas were absent and a special
stain for fungal organisms was negative. There was no evidence of
malignancy. With immunohistochemistry, immunoglobulin (Ig) G4–positive
cells constituted an average of 60 cells per high-power field
(magnification, x 40) and the ratio of IgG4:IgG positive cells was more
than 40% (Figs 2c and 2d). The predominance of IgG4-positive
cells in the lesions excluded ordinary chronic inflammatory changes.
Immunostains for dendritic cell markers (CD1a and CR2) and S-100
proteins were negative, ruling out Langerhans cell histiocytosis. The
bone specimen showed a moderate infiltrate of plasma cells and
lymphocytes, with some polymorphs in the marrow spaces. Immunostaining
of the bone specimen showed a range of 12 to 27 IgG4-positive cells per
high-power field. The overall features were suggestive of IgG4-related
disease (IgG4-RD).
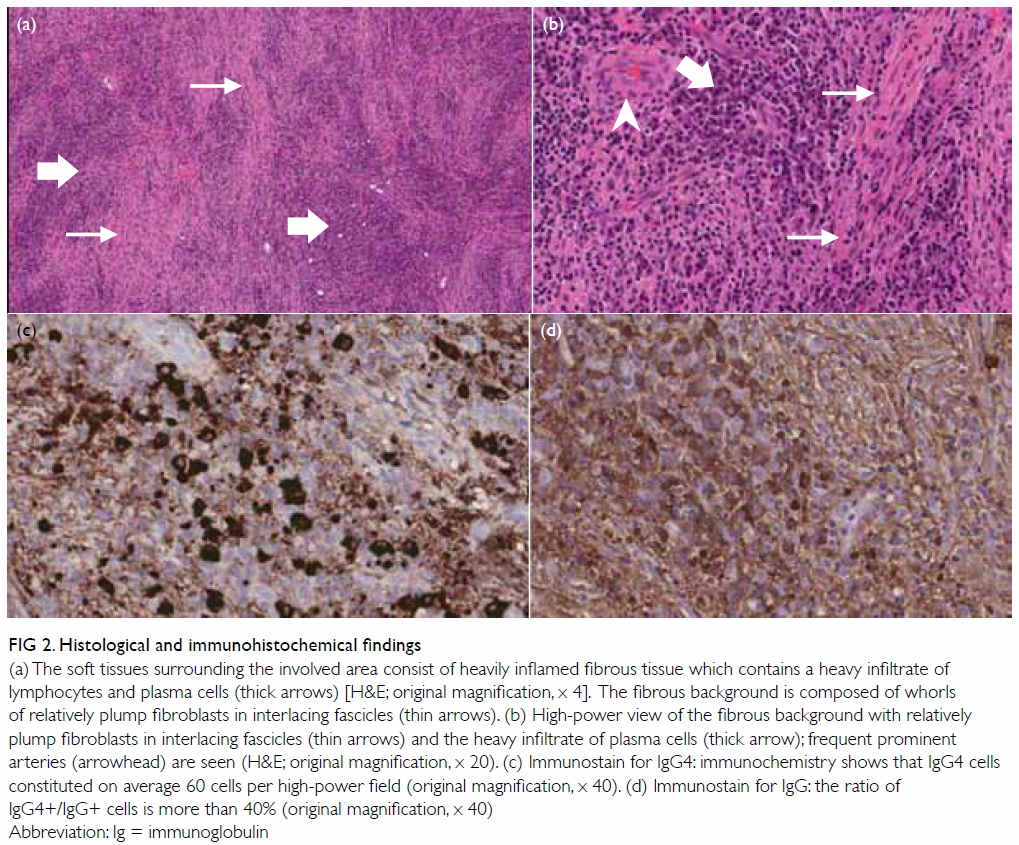
Figure 2. Histological and immunohistochemical findings
(a) The soft tissues surrounding the involved area consist of heavily inflamed fibrous tissue which contains a heavy infiltrate of lymphocytes and plasma cells (thick arrows) [H&E; original magnification, x 4]. The fibrous background is composed of whorls of relatively plump fibroblasts in interlacing fascicles (thin arrows). (b) High-power view of the fibrous background with relatively plump fibroblasts in interlacing fascicles (thin arrows) and the heavy infiltrate of plasma cells (thick arrow); frequent prominent arteries (arrowhead) are seen (H&E; original magnification, x 20). (c) Immunostain for IgG4: immunochemistry shows that IgG4 cells constituted on average 60 cells per high-power field (original magnification, x 40). (d) Immunostain for IgG: the ratio of IgG4+/IgG+ cells is more than 40% (original magnification, x 40)
Abbreviation: Ig = immunoglobulin
Complete blood picture, and liver and renal
function tests were all normal. Serum IgG4 level was within the normal
range. Her IgG level was 1410 mg/dL (reference range, 919-1725 mg/dL),
IgA 272 mg/dL (70-386 mg/dL), IgM 162 mg/dL (55-307 mg/dL), and IgG4
0.88 g/L (0.030-2.0 g/L). Systemic involvement of IgG4-RD was not
evident on clinical examination or positron emission tomography–computed tomography (PET-CT) from skull base to thighs. Systemic
steroid treatment was commenced with dexamethasone 8 mg daily. The
patient reported symptomatic improvements 1 week after steroid
treatment. Mobility of the lower right canine tooth decreased and
returned to normal 3 weeks after steroid treatment.
The patient was also referred to a
rheumatologist. Systemic involvement of IgG4 was not found. Four weeks
after initiation of steroid medication, dexamethasone was replaced
with prednisolone 40 mg daily and maintained for another 4 weeks.
After 8 weeks of systemic steroid administration, the dosage of
prednisolone was reduced by 5 mg each week. Concurrently, azathioprine
25 mg per day was added with increments of 25 mg per week to reach a
dosage of 100 mg per day. Eight months after initiation of medical
treatment, the patient was maintained on azathioprine 100 mg per day
and prednisolone 5 mg on alternate days. There was no evidence of
recurrent clinical signs or symptoms 18 months after initiation of
medical treatment.
Discussion
The IgG4-RD, also called IgG4-related
sclerosing disease, is a newly recognised clinicopathological entity
characterised by intense fibrosis and lymphoplasmacytic infiltration
of involved tissues.1 The
lesions show increased numbers of IgG4-positive plasma cells and are
usually associated with a raised serum IgG4 level. Classically,
IgG4-RD involves the pancreas, hepatobiliary tract, major salivary
glands (including the lacrimal glands), lymph nodes, orbit, and lungs.
In the maxillofacial region, IgG4-RD usually
involves the salivary, lacrimal, and pituitary glands. In the recent
literature, it has also been reported to involve the nasal cavity,
tongue, palatine tonsil, and hard palate.2
To the best of our knowledge, IgG4-RD has not been reported in the
tooth-bearing region of the maxilla or the mandible. Characteristic
organ involvement, elevated serum IgG4 levels, typical histopathology,
and immunohistochemistry are supportive of a diagnosis of IgG4-RD.
Criteria for the diagnosis of IgG4-RD in various organs have been
proposed by Umehara et al3 and
are summarised in the Box.

Interestingly, the present case had a normal
serum IgG4 level. In a previous retrospective analysis of 12 cases
of IgG4-RD in Hong Kong, eight showed raised serum IgG4 levels.4 The pancreas, biliary tract, cervical lymph
nodes, and salivary glands were the involved sites. None of the
cases involved the oral cavity.
There is no consensus or approved treatment
for IgG4-RD. Corticosteroids are the mainstay of therapy. Of note,
PET-CT may be useful in defining disease extent and organ
involvement more accurately. There are no prospective data to
support the starting dose, tapering regimen, or duration of steroid
treatment.
Khurram et al5
used a regimen of dexamethasone 10 mg daily that was reduced to 6 mg
after a week. Dexamethasone was then replaced by prednisolone 5 mg
for a month followed by hydrocortisone pellets for maintenance. The
Japanese strategy consists of treatment with prednisolone (0.6
mg/kg) for 2 to 4 weeks, tapered over 3 to 6 months to 5 mg daily.6 This is followed by
maintenance therapy with 2.5 to 5 mg steroids for 6 months to 3
years. Remission rates are higher than 95%.
Serum IgG4 levels are not reliable for
monitoring therapy response or predicting disease relapse, and may
not be raised in IgG4-RD. Raised serum IgG4 levels can remain
elevated in over half of patients following treatment. Most relapses
occur in the first 3 years following diagnosis and are
steroid-responsive. Relapses can involve different organs to those at
the initial presentation.6
References
1. Cheuk W, Chan JK. IgG4-related
sclerosing disease: a critical appraisal of an evolving
clinicopathologic entity. Adv Anat Pathol 2010;17:303-32. Crossref
2. Andrew N, Kearney D, Sladden N, Goss
A, Selva D. Immunoglobulin G4-related disease of the hard palate. J
Oral Maxillofac Surg 2014;72:717-23. Crossref
3. Umehara H, Okazaki K, Masaki Y, et
al. Comprehensive diagnostic criteria for IgG4-related disease
(IgG4-RD), 2011. Mod Rheumatol 2012;22:21-30. Crossref
4. Ng TL, Leong IS, Tang WL, et al.
Immunoglobulin G4-related sclerosing disease: experience with this
novel entity in a local hospital. Hong Kong Med J 2011;17:280-5.
5. Khurram SA, Fernando M, Smith AT,
Hunter KD. IgG4-related sclerosing disease clinically mimicking
oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral
Radiol 2013;115:e48-51. Crossref
6. Kamisawa T, Okazaki K, Kawa S,
Shimosegawa T, Tanaka M; Research Committee for Intractable
Pancreatic Disease and Japan Pancreas Society. Japanese consensus
guidelines for management of autoimmune pancreatitis: III. Treatment
and prognosis of AIP. J Gastroenterol 2010;45:471-7. Crossref