DOI: 10.12809/hkmj154579
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
A common PRRT2 mutation in familial
paroxysmal kinesigenic dyskinesia in Hong Kong:
a case series of 16 patients
CY Law, PhD, FHKAM (Pathology)1 #; WL Yeung, FHKAM (Paediatrics)2 #; YF Cheung, MPH, FRCP3 #; HF Chan, FHKAM (Medicine)3; Eva Fung, FHKAM (Paediatrics)4; Joannie Hui, FHKAM (Paediatrics)4; Iris OK Yung, MD5; YP Yuen, MSc, FHKAM (Pathology)6; Angel OK Chan, MD, FHKAM (Pathology)7; CW Lam, PhD, FHKAM (Pathology)1
1 Department of Pathology, The University of Hong Kong, Pokfulam, Hong Kong
2 Department of Paediatrics and Adolescent Medicine, Alice Ho Miu Ling Nethersole Hospital, Tai Po, Hong Kong
3 Division of Neurology, Department of Medicine, Queen Elizabeth Hospital, Jordan, Hong Kong
4 Department of Paediatrics, Prince of Wales Hospital, Shatin, Hong Kong
5 Division of Neurology, Department of Medicine and Therapeutics, Prince of Wales Hospital, Shatin, Hong Kong
6 Department of Chemical Pathology, Prince of Wales Hospital, Shatin, Hong Kong
7 Division of Clinical Biochemistry, Queen Mary Hospital, Pokfulam, Hong Kong
# These authors contributed equally to this work.
Corresponding author: Dr CW Lam (ching-wanlam@pathology.hku.hk)
 Full
paper in PDF
Full
paper in PDF
Case reports
Family 1
Patient 1 was a 47-year-old woman who presented
with paroxysmal abnormal movement since
childhood. She was first seen in 2004. The attack
was characterised by dystonic painful posture
with upward or downward deviation of the eyes,
head turning, shoulder abduction and extensions,
lower limb dystonia, impaired speech, and
occasionally stepping movements, with preserved
consciousness. The attacks lasted for 1 to 2 minutes
with a frequency of up to 8 times a day. Each attack
was precipitated by stress or being startled, for
example, being approached by a car unexpectedly
when crossing the road, frightened by a cockroach,
a sudden phone call, or an abrupt movement.
Occasionally, the attack could occur during sleep.
There was no incontinence. The patient had normal
intelligence. Physical examination, biochemical
tests, computed tomography of the brain, and
electroencephalography were normal. The attacks
were controlled with carbamazepine and frequency
of attacks reduced to fewer than 3 times a day and
each episode was shortened to 10 to 20 seconds.
The patient also had a history of seizure of unknown
aetiology in childhood. She had a strong family
history; her daughter has presented with seizure on
two to three occasions since the age of 10 months
and had been taking an anticonvulsant for 2 years.
Since then she had experienced no further seizures
and by the age of 4 years no antiepileptic drug or
follow-up has been required. Two older sisters and a
nephew of the proband also had childhood seizures.
Family 2
Patient 2 was a 26-year-old woman who presented
with paroxysmal kinesigenic dyskinesia (PKD) since
the age of 8 years. The patient first presented with
episodes of afebrile seizure at 7 months old in 1997.
She was prescribed phenobarbitone and required
no further medication or follow-up after the age of
3 years. The current involuntary movement affected
all limbs with the right side being most affected.
Each attack lasted for a few seconds to less than 3
minutes without loss of consciousness. Each episode
was triggered by sudden body movement, anxiety,
or deprived sleep. Frequency of attacks could reach
up to 6 times a day. The patient claimed she could
occasionally partially control the symptoms. Physical
examination, biochemical tests, and brain imaging
were unremarkable. Her brother was also affected by
PKD. The attack was controlled by carbamazepine; a
missed dose would usually lead to relapse.
Family 3
Patient 3 was a 14-year-old boy who presented in
2004 with chorea-like movement of the four limbs
since the age of 18 months. Each attack lasted for
about 15 to 20 seconds and there was no impaired
consciousness. The frequency of attacks could reach
up to 10 times a day. The attacks were triggered after
running or waking up from sleep. A good clinical
response was achieved with carbamazepine. The
attacks recurred only after a missed dose. Physical
examination, biochemical investigations, and brain
imaging were unremarkable and he had normal
development and intelligence. Five paternal family
members were similarly affected.
Family 4
Patient 4 was a 33-year-old man who presented
with involuntary movement since the age of 9 years.
He was first seen in 2011. The involuntary body
movement affected his right facial muscle and right
upper and lower limbs. The attack usually lasted for
5 to 10 seconds with no impaired consciousness.
No remarkable triggering factor was noted. Physical
examination, biochemical tests, and brain imaging
were unremarkable. The patient also experienced
migraine triggered by increased stress. He had
been prescribed carbamazepine since the age of
10 years and a missed dose would lead to relapse.
Two of his daughters were affected by seizures.
The elder daughter presented at 7 months old with
two episodes of generalised seizure, each lasting
around 30 seconds. The younger daughter presented
at 3 months old with a brief lip smacking, limb
twitching, and up-rolling eyeball; the attack lasted
about 2 minutes. Both daughters were prescribed
phenobarbitone. They achieved normal growth and
no further antiepileptic drug was required at 34
months of age for the elder sister while the younger
sister continued on low-dose phenobarbitone.
Family 5
Patient 5 was a 14-year-old boy who presented
with PKD since the age of 9 years. He presented
with on-and-off dystonic movement involving the
limbs, trunk, and occasionally the face. The attacks
lasted for less than 10 seconds and there was no
impaired consciousness. Each episode was triggered
by sudden body movement, for example, starting to
write or going out of an elevator. The frequency of
attacks ranged from once every 2 days to twice a day.
Physical examination and biochemical investigations
were unremarkable. Magnetic resonance imaging
of the brain showed an incidental finding of left
temporal arachnoid cyst. A trial of carbamazepine
achieved a good clinical response.
Family 6
Patient 6 was a 29-year-old man who presented with
PKD around the age of 3 years in 1988. He presented
with jerky variable involuntary movement of all limbs
without loss of consciousness. The attacks usually
lasted for less than 1 minute and were triggered
by sudden body movements. The attacks were not
precipitated by alcohol, caffeine, fatigue, stress, or
excitement. The frequency of attacks could be daily.
Aura was noted before dystonic movement. Physical
examination, biochemical tests, and brain imaging
were unremarkable. The attacks were effectively
controlled by carbamazepine. His father had been
similarly affected since his teenage years. Each
attack usually lasted for 1 to 2 minutes, precipitated
by prolonged sitting or excitement. Aura was noted
before dystonic movement. The sister of the proband
was mildly affected with sudden tightness and
stiffness of limbs without impaired consciousness.
Aura was reported before the dystonic movement.
The other three siblings of the proband were
asymptomatic.
Mutational analysis for PRRT2 gene
Exons of PRRT2 were amplified using polymerase
chain reaction, of which its conditions and primer
sequences for PRRT2 are available upon request.
Sequencing results were compared with the National
Center for Biotechnology Information reference
sequences NM_145239.2 and NP_660282.2.
Mutation analysis for PRRT2 gene in families 1
to 5 showed a heterozygous frameshift mutation
c.649dupC (p.Arg217Profs*8) [Fig 1]. This is a known
pathogenic mutation that can result in premature
transcription termination and a truncated PRRT2
protein. For family 1, DNA samples were not
available for other affected family members except
the proband. For family 6, a novel heterozygous
frameshift mutation, c.379delG (p.Glu127Serfs*49),
was detected (Fig 2). The mutation will lead to
premature transcription termination and a truncated
PRRT2 protein.
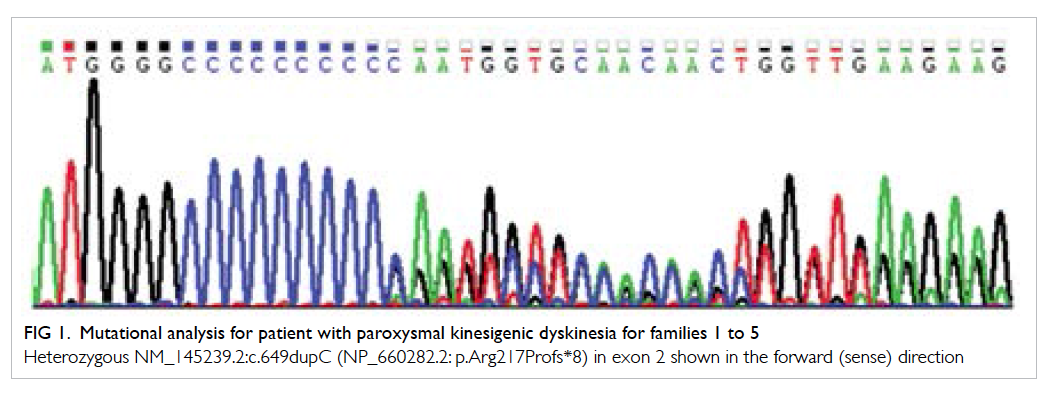
Figure 1. Mutational analysis for patient with paroxysmal kinesigenic dyskinesia for families 1 to 5
Heterozygous NM_145239.2:c.649dupC (NP_660282.2: p.Arg217Profs*8) in exon 2 shown in the forward (sense) direction
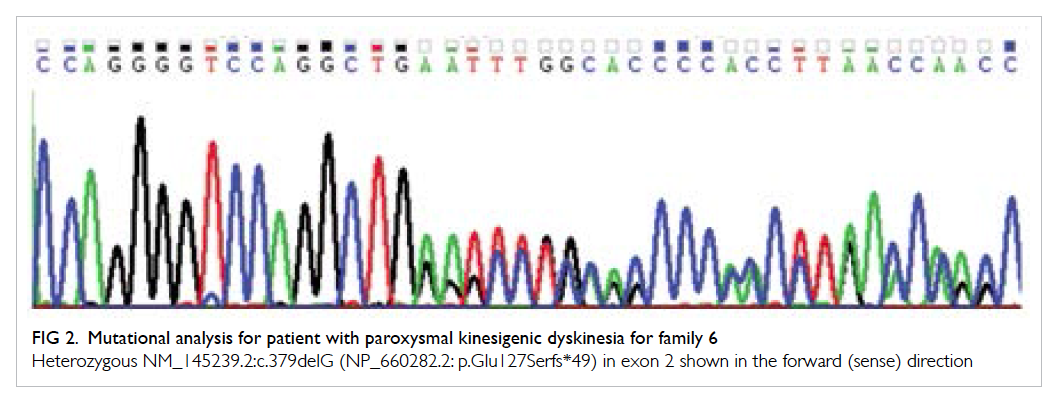
Figure 2. Mutational analysis for patient with paroxysmal kinesigenic dyskinesia for family 6
Heterozygous NM_145239.2:c.379delG (NP_660282.2: p.Glu127Serfs*49) in exon 2 shown in the forward (sense) direction
Discussion
Familial PKD (OMIM#128200) is the most common
type of paroxysmal movement disorder. It is
defined by the presence of (1) identified kinesigenic
triggers, (2) short duration of attacks, usually <1
minute, (3) no loss of consciousness or pain during
attacks, (4) absence of known organic causes and
normal neurological examination, (5) response to
phenytoin or carbamazepine treatment, and (6) age
of onset at 1 to 20 years in the absence of a family
history of PKD.1 The attack is characterised by the
presence of involuntary movement such as dystonia,
choreoathetosis, and ballism. The co-existence of
PKD and infantile convulsions can occur in a subset
of PKD patients and is known as PKD with infantile
convulsions (PKD/IC).
The disorder PKD is transmitted as an
autosomal dominant trait with incomplete
penetrance. The disease is caused by mutation
of the PRRT2 gene (proline-rich transmembrane
protein 2; OMIM*614386).2 3 PRRT2 mutation can also cause PKD/IC, paroxysmal exercise–induced
dyskinesia, paroxysmal non-kinesigenic dyskinesia,
benign familial infantile epilepsy, episodic ataxia,
hemiplegic migraine, intellectual disability, and
benign paroxysmal torticollis of infancy.4 5 Patients with PKD can be misdiagnosed with epilepsy or
psychogenic illness6; an aetiological diagnosis will
require genetic analysis of PRRT2 gene, the most
common disease-causing gene for PKD.
PRRT2 mutation is the major cause of PKD
in Chinese, Koreans, Japanese, and Europeans7 8 9 10 11
and accounts for 33% to 46% of sporadic and 80%
to 100% of familial forms of PKD.12 Heterozygous
c.649dupC is a mutation hotspot of which the
mutation detection rate for Chinese can be as high
as 62%.13 Our findings also suggest that the majority
(81.3%; 95% confidence interval, 57.0%-93.4%) of
PKD patients (13 out of 16) carried the c.649dupC
mutation.
Most (79%) PKD patients had a distinctive
phenotype.1 Atypical features have been reported,
for example, long duration of attack,2 attacks
triggered by stress/anxiety,3 and painful dystonia.14 This explains the unusual features observed in family
1 (painful dystonia, attacks precipitated by stress)
and family 2 (attacks up to 3 minutes). Intriguingly,
no obvious triggering factors were identified in
family 4, likely because they were too subtle and not
recognised by the patient.
Co-existence of infantile convulsion can occur
in a subset of PKD patients (ie PKD/IC), as with the
proband in families 1 and 2. Nevertheless, PKD was
not apparent in the affected children of families 1
and 4, likely because the mean age of onset was 11.6 ± 3.5 years,1 and they were too young for PKD manifestation.
An aetiological diagnosis for patients with PKD
is clinically important. First, the majority of patients
show an excellent response to carbamazepine or
phenytoin and some show significant improvement
with low-dose carbamazepine.15 Second,
neurologists, paediatricians, and pathologists can
explain to the family the aetiology of PKD. Third,
patients can be misdiagnosed with epilepsy or
‘psychogenic illness’. A definitive diagnosis can
end the diagnostic uncertainty and relieve patients
of the emotional uncertainty and unnecessary
investigations.
In conclusion, the cases reported here
constitute the first genetic-confirmed series of PKD
in Hong Kong. We recommend PRRT2 c.649dupC
screening for all patients with all forms of PKD.
References
1. Bruno MK, Hallett M, Gwinn-Hardy K, et al. Clinical
evaluation of idiopathic paroxysmal kinesigenic
dyskinesia: new diagnostic criteria. Neurology
2004;63:2280-7. Crossref
2. Chen WJ, Lin Y, Xiong ZQ, et al. Exome sequencing identifies
truncating mutations in PRRT2 that cause paroxysmal
kinesigenic dyskinesia. Nat Genet 2011;43:1252-5. Crossref
3. Wang JL, Cao L, Li XH, et al. Identification of PRRT2 as
the causative gene of paroxysmal kinesigenic dyskinesias.
Brain 2011;134:3493-501. Crossref
4. Liu Q, Qi Z, Wan XH, et al. Mutations in PRRT2 result in
paroxysmal dyskinesias with marked variability in clinical
expression. J Med Genet 2012;49:79-82. Crossref
5. Gardiner AR, Bhatia KP, Stamelou M, et al. PRRT2 gene
mutations: from paroxysmal dyskinesia to episodic ataxia
and hemiplegic migraine. Neurology 2012;79:2115-21. Crossref
6. Matsumoto N, Takahashi S, Okayama A, Araki A, Azuma
H. Benign infantile convulsion as a diagnostic clue of
paroxysmal kinesigenic dyskinesia: a case series. J Med
Case Rep 2014;8:174. Crossref
7. Chen YP, Song W, Yang J, et al. PRRT2 mutation screening
in patients with paroxysmal kinesigenic dyskinesia from
Southwest China. Eur J Neurol 2014;21:174-6. Crossref
8. Youn J, Kim JS, Lee M, et al. Clinical manifestations in
paroxysmal kinesigenic dyskinesia patients with proline-rich
transmembrane protein 2 gene mutation. J Clin
Neurol 2014;10:50-4. Crossref
9. Okumura A, Shimojima K, Kubota T, et al. PRRT2 mutation
in Japanese children with benign infantile epilepsy. Brain
Dev 2013;35:641-6. Crossref
10. Méneret A, Grabli D, Depienne C, et al. PRRT2 mutations:
a major cause of paroxysmal kinesigenic dyskinesia in the
European population. Neurology 2012;79:170-4. Crossref
11. Becker F, Schubert J, Striano P, et al. PRRT2-related
disorders: further PKD and ICCA cases and review of the
literature. J Neurol 2013;260:1234-44. Crossref
12. Labate A, Tarantino P, Viri M, et al. Homozygous
c.649dupC mutation in PRRT2 worsens the BFIS/PKD
phenotype with mental retardation, episodic ataxia, and
absences. Epilepsia 2012;53:e196-9. Crossref
13. Cao L, Huang XJ, Zheng L, Xiao Q, Wang XJ, Chen SD.
Identification of a novel PRRT2 mutation in patients
with paroxysmal kinesigenic dyskinesias and c.649dupC
as a mutation hot-spot. Parkinsonism Relat Disord
2012;18:704-6. Crossref
14. Ebrahimi-Fakhari D, Kang KS, Kotzaeridou U, Kohlhase J,
Klein C, Assmann BE. Child Neurology: PRRT2-associated
movement disorders and differential diagnoses. Neurology
2014;83:1680-3. Crossref
15. Chou IC, Lin SS, Lin WD, et al. Successful control with
carbamazepine of family with paroxysmal kinesigenic
dyskinesia of PRRT2 mutation. Biomedicine (Taipei)
2014;4:15. Crossref