Hong Kong Med J 2016 Aug;22(4):314–9 | Epub 3 Jun 2016
DOI: 10.12809/hkmj154653
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Clinical and genetic profile of catecholaminergic polymorphic ventricular tachycardia in Hong Kong Chinese children
TC Yu, MB, ChB, FHKAM (Paediatrics)1;
Anthony PY Liu, MB, BS, MRCPCH2;
KS Lun, MB, ChB, FHKAM (Paediatrics)3;
Brian HY Chung, MB, ChB, FHKAM (Paediatrics)2;
TC Yung, MB, BS, FHKAM (Paediatrics)3
1 Department of Paediatrics and Adolescent Medicine, Pamela Youde
Nethersole Eastern Hospital, Chai Wan, Hong Kong
2 Department of Paediatrics and Adolescent Medicine, Li Ka Shing Faculty of
Medicine, The University of Hong Kong, Pokfulam, Hong Kong
3 Department of Paediatric Cardiology, Queen Mary Hospital, Pokfulam, Hong Kong
Corresponding author: Dr TC Yu (ytc604@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Objective: To report our experience in the
management of catecholaminergic polymorphic
ventricular tachycardia in Hong Kong Chinese
children.
Methods: This case series study was conducted in a
tertiary paediatric cardiology centre in Hong Kong.
All paediatric patients diagnosed at our centre
with catecholaminergic polymorphic ventricular
tachycardia from January 2008 to October 2014 were
included.
Results: Ten patients (five females and five males)
were identified. The mean age at presentation
and at diagnosis were 11.0 (standard deviation,
2.9) years and 12.5 (2.8) years, respectively. The
mean delay time from first presentation to diagnosis was 1.5
(standard deviation, 1.3) years. They presented with
recurrent syncope and six patients had a history of
aborted cardiac arrest. Four patients were initially
misdiagnosed to have epilepsy. Catecholaminergic
polymorphic ventricular tachycardia was diagnosed
by electrocardiogram at cardiac arrest (n=2), or
provocation test, either by catecholamine infusion
test (n=6) or exercise test (n=2). Mutations of the RyR2
gene were confirmed in six patients. Nine patients
were commenced on beta-blockers after diagnosis.
Despite medications, three patients developed
aborted or resuscitated cardiac arrest (n=2) and
syncope (n=1). Left cardiac sympathetic denervation
was performed in five patients and an implantable
cardioverter defibrillator was implanted in another.
There was no mortality during follow-up.
Conclusions: Catecholaminergic polymorphic
ventricular tachycardia should be considered in
children who present with recurrent syncope during
exercise or emotional stress. Despite beta-blocker
treatment, recurrent ventricular arrhythmias occur
and may result in cardiac arrest.
New knowledge added by this study
- This is the first study of catecholaminergic polymorphic ventricular tachycardia (CPVT) in Hong Kong describing local experience in the management of this rare arrhythmic syndrome.
- The genetic background (RyR2 mutation) of our Chinese children is similar to those in overseas studies.
- CPVT should be considered in young patients who present with exercise-related syncope.
- Maintaining a high index of suspicion and correct diagnosis of CPVT may be life-saving.
Introduction
Catecholaminergic polymorphic ventricular
tachycardia (CPVT) is an inherited arrhythmia
syndrome. Mutation of the ryanodine receptor
2 (RyR2) gene and infrequently the calsequestrin
(CASQ2) gene is identified in approximately 60%
to 70% of patients.1 2 Patients with CPVT usually present with syncope and sudden cardiac death.
The symptoms are due to bidirectional polymorphic
ventricular tachycardia (VT) induced by adrenergic
stress.1 Onset of arrhythmia syndrome is usually in
childhood. Many affected children are considered to
have vasovagal syncope or epilepsy before a correct
diagnosis is made.1 2 3 4 If left untreated, the mortality of
CPVT is up to 31% by the age of 30 years.1 3 5
In this study, we reviewed the clinical
characteristics, genetic profile, and outcome of
CPVT in Hong Kong Chinese children.
Methods
Our study included children diagnosed with CPVT
from January 2008 to October 2014 at Queen
Mary Hospital, a university-affiliated teaching
hospital in Hong Kong. The hospital records were
retrospectively reviewed. Demographic data, clinical
presentation, diagnostic methods, and genetic tests
were reviewed. In all patients, the heart rate–corrected QT interval of
the resting electrocardiogram was normal and the
presence of structural heart disease was excluded
by echocardiography (n=10) and/or magnetic
resonance imaging (n=5). We also summarised the
treatment modalities, response to treatment, and
clinical outcome up to October 2014.
Genetic analysis
Blood samples of seven patients were sent to
the Molecular Genetics Laboratory of Victorian
Clinical Genetic Services, Australia where testing for
mutations of the RyR2 gene was performed. The assay
involved sequencing of 17 hotspot exons (exons 1,
8, 14, 15, 44, 46, 47, 49, 88, 93, 95, 97, 101, 102, 103,
104, 105), their splice junctions and 20 bps into the
introns. Since 2014, the Laboratory has made use
of a cardiac next-generation sequencing panel to
analyse the 28 arrhythmia genes: AKAP9, ANK2,
CACNA1C, CACNA2D1, CACNB2, CASQ2, CAV3,
GJA5, GPD1L, HCN4, KCNA5, KCND3, KCNE1,
KCNE1L, KCNE2, KCNE3, KCNH2, KCNJ2, KCNJ5,
KCNJ8, KCNQ1, NPPA, RYR2, SCN1B, SCN3B,
SCN4B, SCN5A, and SNTA1. In two patients, the
samples were tested by the local Laboratory Genetic
Service (Department of Pathology, Princess Margaret
Hospital, Hong Kong), where direct sequencing of
selected hotspot exons and the flanking introns (10
bps) was performed. Cascade testing was offered for
first-degree relatives of genotype-positive subjects.
Results
Characteristics of the study subjects
During the study period, 10 patients were diagnosed
to have CPVT. Their demographic data and clinical
features are summarised in Table 1. The group
comprised five female and five male patients; two
of whom were brothers. The mean (± standard
deviation) age at first presentation was 11.0 ± 2.9
(range, 6.2-14.2) years. The mean age at diagnosis
was 12.5 ± 2.8 (range, 6.9-15.1) years. The mean delay time from
first presentation to diagnosis was 1.5 ± 1.3 years.
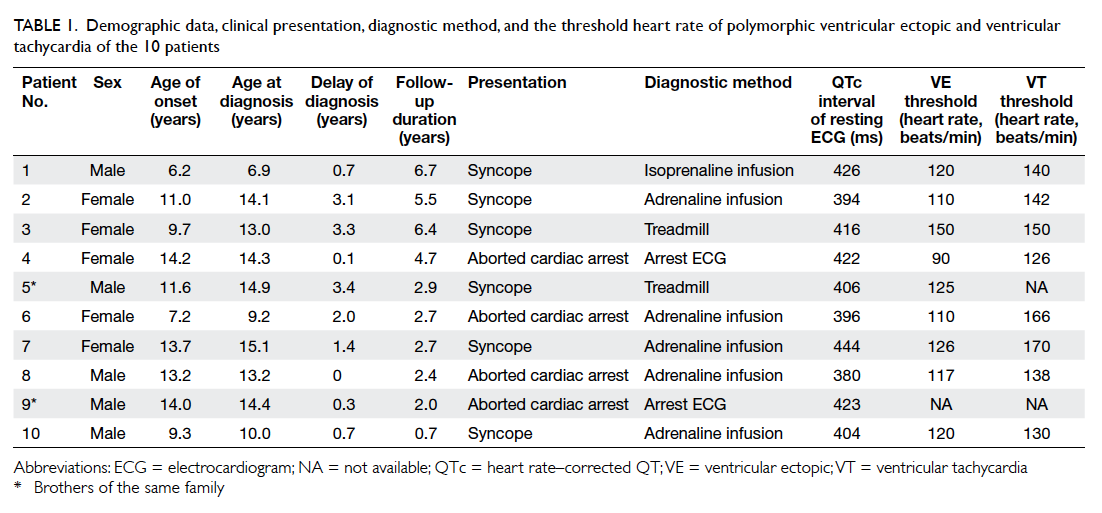
Table 1. Demographic data, clinical presentation, diagnostic method, and the threshold heart rate of polymorphic ventricular ectopic and ventricular tachycardia of the 10 patients
Six patients presented initially with syncope
while the other four presented with aborted cardiac
arrest. At the end of the study, a total of six patients
had aborted cardiac arrest. The triggering event
for syncope or cardiac arrest was either exercise or
emotion. Nonetheless, no such event was evident in
three patients.
Four patients were initially misdiagnosed
with epilepsy, one of whom was treated with an
anticonvulsant prior to the diagnosis of CPVT.
Of the four patients who presented with
aborted cardiac arrest, three required repeated
cardioversion because of recurrent VT immediately
following successful termination of ventricular
arrhythmias. The case of patient 4 has been reported
previously.6
Diagnosis of catecholaminergic polymorphic ventricular tachycardia and genetic analysis
Diagnosis of CPVT in two patients was based on
the presence of bidirectional polymorphic VT in the
cardiac arrest electrocardiogram. In the remaining
patients, diagnosis was made when polymorphic or
bidirectional VT was induced during provocation
tests by exercise (n=2) or catecholamine infusion (n=6). Heart
rate at the induction of ventricular premature beats
ranged from 90 to 150 beats/min. Polymorphic VTs
were induced when heart rate was increased to 126
to 170 beats/min (Fig).
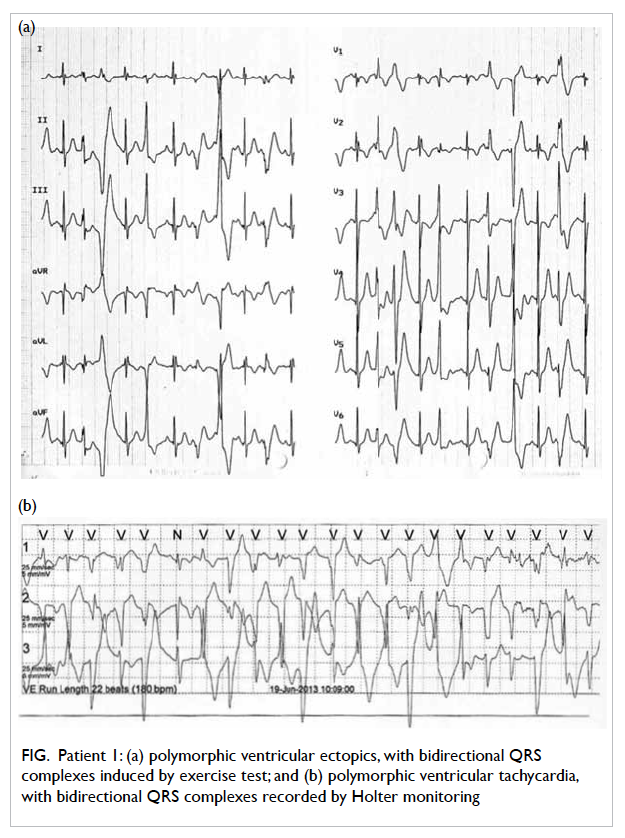
Figure. Patient 1: (a) polymorphic ventricular ectopics, with bidirectional QRS complexes induced by exercise test; and (b) polymorphic ventricular tachycardia, with bidirectional QRS complexes recorded by Holter monitoring
Of the nine patients with genetic study, six
were confirmed to have mutations of the RyR2 gene
as shown in Table 2. One patient (patient 9) did not
undergo genetic study because his brother (patient
5) was confirmed to have no mutation of RyR2. Only
two (brothers of the same family) of 10 patients had
a family history of cardiac arrhythmic events. There
was no RyR2 mutation identified in the first-degree
relatives of any patient with a RyR2 mutation.
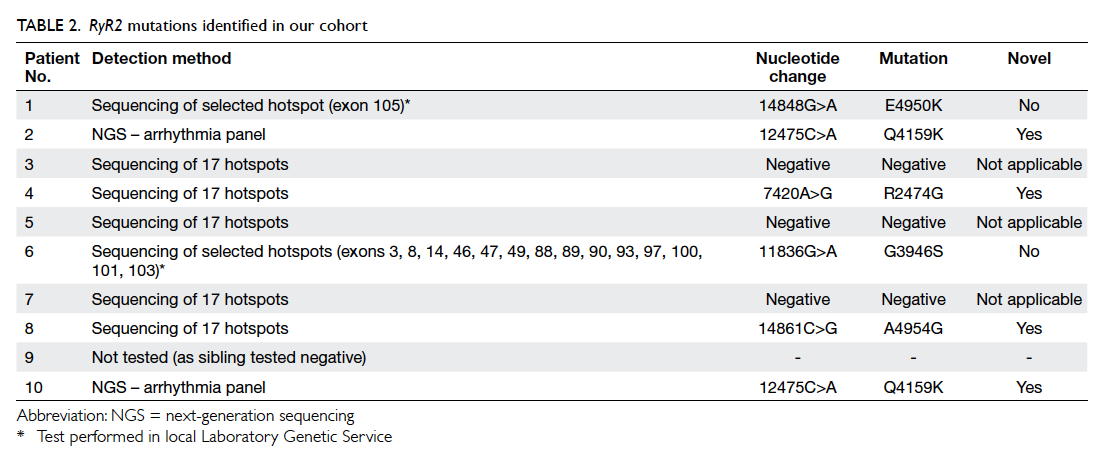
Table 2. RyR2 mutations identified in our cohort
Treatment and response
Medical treatment
The treatment modalities and response are
summarised in Table 3. All patients were started
on a beta-blocker as first-line medication. One
patient initially refused medical treatment. She then
had recurrent syncope and subsequently agreed to
treatment with nadolol.
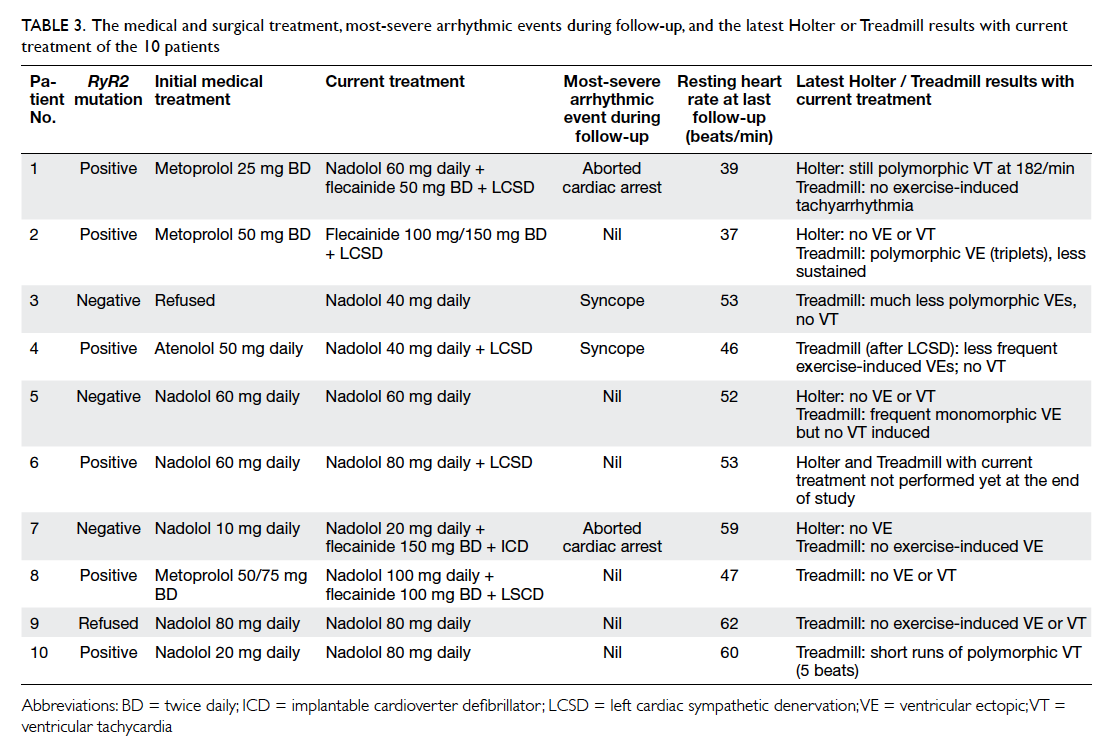
Table 3. The medical and surgical treatment, most-severe arrhythmic events during follow-up, and the latest Holter or Treadmill results with current treatment of the 10 patients
Metoprolol was prescribed to three patients
as initial medical treatment, although all switched
to nadolol with or without flecainide due to
unsatisfactory control (aborted cardiac arrest in one
and exercise-induced polymorphic VT in another)
or intolerable side-effects (tiredness and significant
bradycardia at 38 beats/min).
Of the six patients prescribed nadolol as the
first medication, five had no more syncope and
no VT on treadmill exercise testing. Nadolol was
changed to flecainide in one patient (patient 7) due
to significant resting bradycardia of 35 beats/min.
Nadolol was later resumed at a lower dose.
Atenolol was started in one girl as initial
medical treatment but failed to prevent recurrent
syncope. After changing to nadolol, she remained
symptomatic and subsequently underwent left
cardiac sympathetic denervation (LCSD).
Additional treatments
Left cardiac sympathetic denervation was performed
via a video-assisted thoracoscopic approach in five
patients. The lower half of the stellate ganglion and
the sympathetic trunk of T2 to T4 were resected.
After LCSD, one patient (patient 1) still had recurrent
syncope. The other four patients had no more
syncope. Dual-chamber implantable cardioverter defibrillator
(ICD) implantation was performed
in one patient (patient 7) who experienced an
aborted cardiac arrest despite flecainide. She had
no complications related to the ICD implantation.
After implantation, she had one episode of syncope
while she was swimming slowly in the pool with her
mother. She was taken out of the water and was able
to stand unaided soon after. The ICD interrogation
noted an episode of VT/ventricular fibrillation that
was successfully aborted by electric shocks from the
ICD. She had no inappropriate shocks from the ICD
during the follow-up period of 30 months.
Outcomes
The median duration of follow-up was 3.7 ± 2.0
(range, 0.7-6.7) years. Six (60%) patients became
asymptomatic after drug treatment. Two patients
had recurrent syncope; one of whom was without
drug treatment. Two patients experienced aborted
cardiac arrest, one received ICD implantation and
another one refused. There was no mortality during
the study period.
Discussion
Catecholaminergic polymorphic ventricular
tachycardia is uncommon in Hong Kong Chinese
children. Our centre treated most of the serious local
paediatric cardiac arrhythmia cases. Over a period
of 7 years we identified only 10 patients. Our case
series is, to date, the largest in Chinese children.
Many of our patients (6 out of 10) had
experienced aborted cardiac arrest as the near-fatal
arrhythmic event during the study. The
diagnosis of CPVT can be challenging and requires
documentation of typical bidirectional polymorphic
VT at presentation, or induction of polymorphic VT
by exercise test or catecholamine infusion test.1 2 3 7 8
Studies show that diagnosis of CPVT can be made
in 69% and 75% of patients by exercise test and
catecholamine infusion test, respectively.9 10
Misdiagnosis and delay in diagnosis of CPVT
is common. Our patients had a mean delay of
1.5 years from first presentation to diagnosis. Four of
our patients were initially misdiagnosed with epilepsy,
one of whom was prescribed anticonvulsant therapy.
Of the 10 patients, four were not diagnosed until
they presented with aborted cardiac arrest.
Genetic mutations are identified in 60% to
70% of patients with CPVT, and more than 90%
of the mutations affect the RyR2 gene.1 3 Mutation of the CASQ2 gene is rare (<2%). Very recently,
mutation of triadin, a transmembrane sarcoplasmic
reticulum protein, was found to be the cause of
CPVT in two families.11 In these mutations, the
defective proteins cause excessive calcium release
from the sarcoplasmic reticulum to the cytoplasm
leading to polymorphic VT.1 5 Similar to overseas studies, mutation of the RyR2 gene was evident in
the majority (60%) of our patients.
Patients with CPVT must be restricted from
exercise to avoid the adrenergic trigger. A beta-blocker
serves as first-line medical therapy.1 2 4 10
Nonetheless, CPVT is a very malignant arrhythmic
disease and many patients remain symptomatic
despite such therapy.1 3 4 10 In a systematic analysis of
354 CPVT patients treated with beta-blockers, the
estimated 8-year arrhythmic event rate was 37.2%.12
Our study also showed that a high proportion of
patients still developed arrhythmic events despite
beta-blocker treatment (syncope in one and aborted
cardiac arrest in two out of 10 patients).
In the early period of study, we prescribed
metoprolol in three patients, although all experienced
treatment failure due to recurrent symptoms or
intolerance. In the later period, nadolol was the
initial medication and five out of six patients became
asymptomatic.
Flecainide, a class 1c anti-arrhythmic drug with
dual action of direct ryanodine receptor blockage
and blockage of sodium channels,1 12 may be effective in CPVT patients. Flecainide has been evaluated in a
multicentre study of 33 CPVT patients. In 22 (76%) out of 29 patients, flecainide suppressed exercise-induced
ventricular arrhythmia either partially (n=8) or
completely (n=14).1 12 13 In our study, flecainide was
used in four patients who had failed treatment with
a beta-blocker. Three patients still had arrhythmic
events, however.
Studies showed that LCSD, which prevents
noradrenaline release in the heart, is highly effective
in severely affected CPVT patients.14 15 It can be performed with a minimally invasive approach by
video-assisted thoracic surgery. In our study, five
patients underwent LCSD. All recovered well and no
complications were noted at follow-up. Four had no
more syncope. Large studies are needed to further
evaluate its efficacy in CPVT patients.
An ICD has been recommended in patients
who fail optimised medical therapy.1 12 14 16 Some
recent studies have suggested that ICD may be
harmful to CPVT patients, however, because both
appropriate and inappropriate ICD shocks can
potentially induce VT storms and cardiac arrest.16 17 Therefore, ICD implantation should be restricted
to patients with symptoms refractory to optimised
medical treatment and LCSD.18
Conclusions
Catecholaminergic polymorphic ventricular
tachycardia is an uncommon but malignant cardiac
arrhythmia that presents as syncope, seizure, or
sudden cardiac death in childhood. In our study,
60% of patients experienced aborted cardiac arrest.
One should suspect the diagnosis when syncope
occurs during exercise or emotional stress. Similar
to overseas studies, RyR2 mutation is the most
common genetic mutation and affected 60% of our
patients. Despite optimised medical therapy, 60% of
patients still required LCSD or ICD implantation.
Acknowledgements
The expenses of genetic analysis were sponsored by
the Children’s Heart Foundation of Hong Kong.
Declaration
All authors have no relevant conflicts of interest to
disclose.
References
1. Ylänen K, Poutanen T, Hiippala A, Swan H, Korppi M.
Catecholaminergic polymorphic ventricular tachycardia.
Eur J Pediatr 2010;169:535-42. Crossref
2. Priori SG, Napolitano C, Memmi M, et al. Clinical
and molecular characterization of patients with
catecholaminergic polymorphic ventricular tachycardia.
Circulation 2002;106:69-74. Crossref
3. Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel
P. Catecholaminergic polymorphic ventricular tachycardia
in children. A 7-year follow-up of 21 patients. Circulation
1995;91:1512-9. Crossref
4. Çeliker A, Erdoğan I, Karagöz T, Özer S. Clinical experiences
of patients with catecholaminergic polymorphic ventricular
tachycardia. Cardiol Young 2009;19:45-52. Crossref
5. Swan H, Piippo K, Viitasalo M, et al. Arrhythmic disorder
mapped to chromosome 1q42-q43 causes malignant
polymorphic ventricular tachycardia in structurally normal
hearts. J Am Coll Cardiol 1999;34:2035-42. Crossref
6. Kung SW, Yung TC, Chiu WK. Successful resuscitation of
out-of-hospital ventricular fibrillation cardiac arrest in an
adolescent. Hong Kong J Emerg Med 2010;17:482-7.
7. Lahat H, Eldar M, Levy-Nissenbaum E, et al. Autosomal
recessive catecholamine- or exercise-induced polymorphic
ventricular tachycardia: clinical features and assignment
of the disease gene to chromosome 1p13-21. Circulation
2001;103:2822-7. Crossref
8. Postma AV, Denjoy I, Kamblock J, et al. Catecholaminergic
polymorphic ventricular tachycardia: RYR2 mutations,
bradycardia, and follow up of the patients. J Med Genet
2005;42:863-70. Crossref
9. Marjamaa A, Hiippala A, Arrhenius B, et al. Intravenous
epinephrine infusion test in diagnosis of catecholaminergic
polymorphic ventricular tachycardia. J Cardiovasc
Electrophysiol 2012;23:194-9. Crossref
10. Sumitomo N, Harada K, Nagashima M, et al.
Catecholaminergic polymorphic ventricular tachycardia:
electrocardiographic characteristics and optimal
therapeutic strategies to prevent sudden death. Heart
2003;89:66-70. Crossref
11. Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, et
al. Absence of triadin, a protein of the calcium release
complex, is responsible for cardiac arrhythmia with sudden
death in human. Hum Mol Genet 2012;21:2759-67. Crossref
12. van der Werf C, Zwinderman AH, Wilde AA. Therapeutic
approach for patients with catecholaminergic polymorphic
ventricular tachycardia: state of the art and future
developments. Europace 2012;14:175-83. Crossref
13. van der Werf C, Kannankeril PJ, Sacher F, et al. Flecainide
therapy reduces exercise-induced ventricular arrhythmias
in patients with catecholaminergic polymorphic ventricular
tachycardia. J Am Coll Cardiol 2011;57:2244-54. Crossref
14. Wilde AA, Bhuiyan ZA, Crotti L, et al. Left cardiac
sympathetic denervation for catecholaminergic
polymorphic ventricular tachycardia. N Engl J Med
2008;358:2024-9. Crossref
15. De Ferrari GM, Dusi V, Spazzolini C, et al. Clinical
management of catecholaminergic polymorphic
ventricular tachycardia: the role of left cardiac sympathetic
denervation. Circulation 2015;131:2185-93. Crossref
16. Miyake CY, Webster G, Czosek RJ, et al. Efficacy of
implantable cardioverter defibrillators in young patients
with catecholaminergic polymorphic ventricular
tachycardia: success depends on substrate. Circ Arrhythm
Electrophysiol 2013;6:579-87. Crossref
17. Mohamed U, Gollob MH, Gow RM, Krahn AD. Sudden
cardiac death despite an implantable cardioverter-defibrillator
in a young female with catecholaminergic
ventricular tachycardia. Heart Rhythm 2006;3:1486-9. Crossref
18. van der Werf C, Wilde AA. Catecholaminergic polymorphic
ventricular tachycardia: from bench to bedside. Heart
2013;99:497-504. Crossref