Hong Kong Med J 2016 Apr;22(2):171–7 | Epub 14 Mar 2016
DOI: 10.12809/hkmj154634
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
REVIEW ARTICLE CME
A new paradigm of genetic testing for hereditary breast/ovarian cancers
Ava Kwong, FRCS (Edin), PhD;
JW Chen, PhD;
Vivian Y Shin, PhD
Breast Surgery Division, The University of Hong Kong, Pokfulam, Hong Kong
Corresponding author: Dr Ava Kwong (akwong@asiabreastregistry.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Introduction: Genetic risk factors and family history
play an important role in breast cancer development.
This review aimed to summarise the current genetic
testing approach to hereditary breast/ovarian cancer.
Methods: A systematic literature review was
performed by searching the PubMed database.
Publications available online until January 2015 that
addressed issues related to hereditary breast/ovarian
cancer genetic counselling/testing were selected. The
search terms used were “familial breast/ovarian
cancer”, “susceptibility genes”, “genetic counselling”,
and “genetic testing”. The data extracted for this review were
analysed by the authors, with a focus on genetic
testing for hereditary breast/ovarian cancer.
Results: Although a greater proportion of inherited breast/ovarian cancers are due to the BRCA1 and BRCA2
mutations, a number of new genes have emerged as
susceptibility candidates, including rare germline
mutations in high penetrance genes, such as TP53 and
PTEN, and more frequent mutations in moderate/low penetrance genes, such as PALB2, CHEK2 and ATM. Multi-gene testing, if used appropriately, is
generally a more cost- and time-effective method
than single-gene testing, and may increase the
number of patients who can be offered personal
surveillance, risk-reduction options, and testing of
high-risk family members.
Conclusions: Recent advances in molecular genetics
testing have identified a number of susceptibility
genes related to hereditary breast and/or ovarian
cancers other than BRCA1 and BRCA2. The
introduction of multi-gene testing for hereditary
cancer has revolutionised the clinical management
of high-risk patients and their families. Individuals
with hereditary breast/ovarian cancer will benefit
from genetic counselling/testing.
Introduction
Breast cancer is one of the most common cancers
and the second most common leading cause of
cancer-related death among women with 1.67
million new cases diagnosed in 2012 (25% of all
cancers).1 About 39% of these new cases are found in
Asia.1 In the US, women have a 12% lifetime risk of
developing breast cancer including women of young
age. In addition, approximately 1 in 250 women in
their 30s will develop breast cancer in the next 10
years.2 Assessment of an individual’s risk for breast
cancer is complex, and based on different aspects
such as personal lifestyle, environmental exposure,
reproductive influences, and drug use. Genetic
risk factors and family history, however, also play
important roles in breast cancer development. Only
5% to 10% of breast cancer cases are characterised as
hereditary and follow the autosomal dominant
pattern of transmission.3 On the other hand,
15% to 20% of breast cancer cases are familial, referring
to women who have two or more first- or second-degree
relatives with the disease.4 5 6 Hereditary
cancers follow a Mendelian inheritance pattern and
tend to have an earlier age of onset. Familial cancers
do not follow a specific inheritance pattern. Defects
in the BRCA1 and BRCA2 genes are the most well-known
high-risk factors among inherited breast
cancers. Results from genome-wide association
studies have broadened our knowledge over the last
few years about the specific genes that contribute to
familial breast cancer. Other genes such as TP53 and
PTEN have also been identified to be associated with
an increased risk of breast cancer.7 High-risk women
are likely to benefit from genetic testing as there are
now emerging targeted therapies and interventions
that have been shown to improve outcome in
mutation carriers.
Methods
A search of the medical literature was performed to
identify the relevant studies and reviews on genetic
testing for hereditary breast/ovarian cancer. The
PubMed database was searched for publications
available online until January 2015 that address
the related issues; “familial breast/ovarian cancer”,
“susceptibility genes”, “genetic counselling”, and “genetic
testing” were used as the search terms.
High-penetrance genes
BRCA1 and BRCA2
Hereditary breast and ovarian cancer syndrome
(HBOC) refers to a germline mutation in either the
BRCA1 or BRCA2 gene, and individuals who carry
a mutation have an increased risk of developing
cancers. BRCA1 and BRCA2 are tumour-suppressor
genes that code for proteins that help repair damaged
DNA and therefore play vital roles in securing the
stability of the cell’s genetic material. Defects in these
two genes may result in protein with malfunction,
thus DNA damage may not be repaired properly.
As a result, cells are prone to develop genetic
mutations leading to cancer development. Scientists
discovered in the 1990s that BRCA1 and BRCA2 are
breast cancer susceptibility genes.8 9 Women have a 57% to 60% and 49% to 55% lifetime risk of developing breast
cancer if they carry a BRCA1 or BRCA2 mutation,
respectively.10 11 Women with mutations in the BRCA1 cancer susceptibility gene associated with
HBOC have a 39% to 46% risk of developing ovarian
cancer by the age of 70 years while approximately
10% to 27% BRCA2-positive women are at risk.12 13 14 The
result of genetic testing for the BRCA mutation is
important to decisions made about management
of breast cancer. For example, a woman diagnosed
with breast cancer and who harbours the BRCA1
or BRCA2 mutation has a greater risk of developing
a second breast cancer in the contralateral breast,
and this risk is age-related. Women diagnosed with
breast cancer at a younger age have a higher risk of
developing contralateral malignancy compared with
those diagnosed at an older age.15 BRCA1 mutation
carriers tend to have more triple-negative breast
cancer (TNBC), medullary histopathology, somatic
TP53 mutations, higher histological grade, and
present at a younger age compared with women
with sporadic breast cancers. Basal markers such as
cytokeratin (CK14, CK5/6, CK17), osteonectin, and
EGFR are more commonly expressed in BRCA1-positive tumours than in control tumours unselected
for mutation status.16 17 18 The National Comprehensive
Cancer Network (NCCN) annually updates
guidelines with respect to genetic counselling and
testing (www.nccn.org) and the most updated
guidelines recommend it for individuals who meet
the HBOC testing criteria. Guidelines are based on
young age of onset, family history of breast cancer,
specific histological types of breast cancer (TNBC),
ovarian (epithelial and peritoneal), and prostate cancer
(Gleason score ≥7). For details refer to NCCN
guidelines (Genetic/Familial High-Risk Assessment:
Breast and Ovarian), version 1.2016.19
Knowing the mutation status of germline
BRCA1 and BRCA2, patients may be offered
alternative screening and/or therapeutic
interventions (Table 119), including intensive breast
surveillance (magnetic resonance imaging [MRI] of
the breasts in addition to standard breast imaging
such as mammography and ultrasonography),
mastectomy instead of breast conservation surgery,
prophylactic mastectomy and salpingo-opherectomy,
or the prescription of chemopreventive drugs and
more recently the choice of chemotherapy as primary
treatment, for example, carboplatin. A recent study
has shown that treatment with carboplatin produces
no advantage over docetaxel in patients with TNBC,
although those with BRCA1 or BRCA2 mutation
benefited from either drug.20 A number of targeted
therapies, such as poly(ADP-ribose) polymerase inhibitors, have been shown
to be effective in BRCA mutation carriers.21 22 The evolution of sequencing technologies enables parallel
testing of multiple genes, leading to simultaneous
analysis of breast cancer predisposition genes with
either high or intermediate penetration. Multi-gene
panel testing, however, has raised new issues
regarding patient eligibility for gene testing other
than BRCA1 and BRCA2, and more importantly,
interpretation of genetic results.
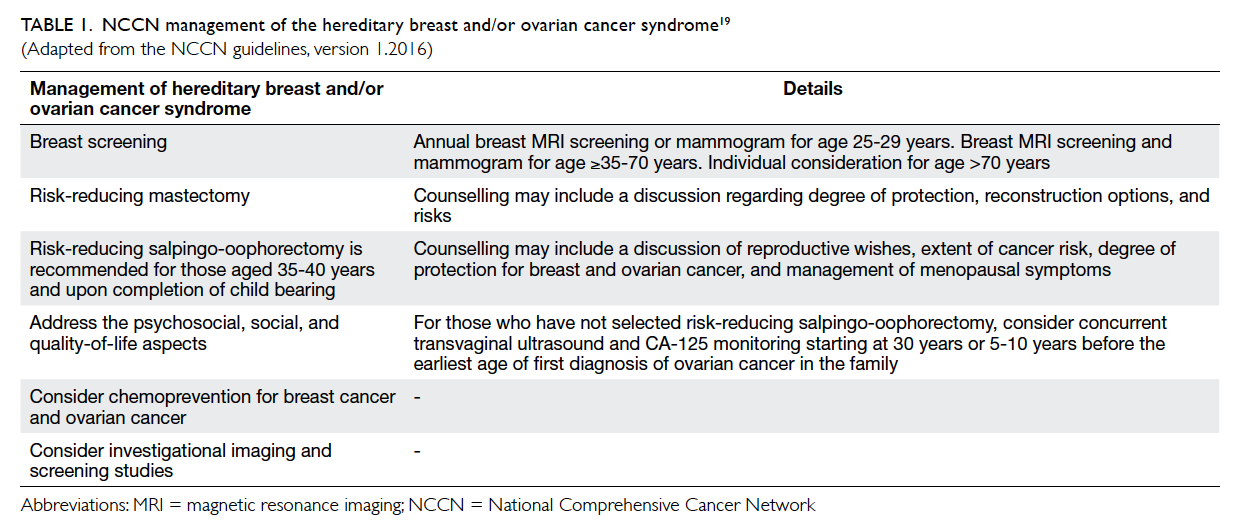
Table 1. NCCN management of the hereditary breast and/or ovarian cancer syndrome19
(Adapted from the NCCN guidelines, version 1.2016)
TP53
One of the high penetrance genes is TP53, which
is a tumour-suppressor gene that encodes the
transcription factor protein p53. It is a ubiquitous
protein implicated in preservation of an intact
genome. It regulates cell cycle, DNA repair,
apoptosis, cellular senescence, and metabolism. It has
been shown to be involved in various kinds of cancer
progression such as osteosarcomas, colon cancer,
and lung cancer.23 24 25 26 27 28 Li-Fraumeni syndrome (LFS) is
a rare but highly penetrance familial cancer syndrome
that is characterised by germline TP53 mutations
inherited in an autosomal dominant manner, in which
60% to 80% of LFS families carry a mutant TP53.29 In
addition to soft-tissue sarcomas and osteosarcomas,
LFS families are likely to exhibit a pattern of early-onset
and multiple primary cancers including breast, brain, and adrenocortical tumours29 30; LFS is thought to account for approximately 1% of all
breast cancers.31 32 Approximately 1% of women diagnosed with breast cancer before the age of 40
years carry a TP53 mutation.32 Breast cancer is the
most frequent malignancy among female TP53
mutation carriers and accounts for up to one third
of all cancers in LFS families.33 Although LFS is only
responsible for a tiny fraction of breast cancers,
women with LFS have a breast cancer risk of 56% by
the age of 45 years and greater than 90% by the age
of 60 years, and LFS accounts for a 60-fold increased
risk for early-onset breast cancer compared with the
general population.34 35 Women with LFS-related breast cancer are reported to have very early disease
onset (20s or 30s) and a relatively advanced disease
staging.36 37 38 Studies have shown that 3% to 8% of women
who are diagnosed with breast cancer younger
than 30 years without a significant family history
of cancer have TP53 mutation.31 39 40 The NCCN
has included early-onset breast cancer as one of the
criteria for offering TP53 genetic testing, regardless
of the family history of cancer. TP53 mutations
can be tested either through sequencing the entire
encoding region that identifies approximately 95%
of TP53 mutations or just selected regions. Analysis
of hot-spot regions located in exons 4-9 can detect
approximately 90% of all TP53 mutations.19 41 42 When
the TP53 mutation is present in an individual, breast
screening and preventive guidelines are similar
to those for BRCA mutation carriers. In addition, a
full-body MRI scan is an option as a screening tool.
Individuals with the following should be included for
genetic testing of TP5319: early-onset breast cancer
(≤35 years), a combination of diagnosis of a sarcoma
at the age of <45 years, AND a first-degree relative
diagnosed at the age of <45 years with cancer,
multiple cancers (brain tumours, sarcomas, and
leukaemia).
PTEN
PTEN is a phosphatase tensin homologue located
on chromosome 10q23.3 that plays a tumour-suppressive
role due to its PI3K (phosphatidylinositol-3-kinase) phosphatase activity. Abnormal PTEN
cannot activate cell cycle arrest and apoptosis and
leads to uncontrolled cell survival.43 Germline PTEN
mutations have been identified in a variety of disorders
such as Cowden syndrome (CS) or PTEN hamartoma
tumour syndrome. Affected individuals have
multiple hamartomas in a variety of tissues with an
increased risk of malignant transformation.44 Breast
cancer is the most common tumour associated with
CS. Although CS is responsible for <1% of all breast
cancers, women with this syndrome have a 25% to 50%
risk of developing breast cancer in a lifetime and are
prone to early onset.45 46 The frequency of multifocal and bilateral disease is increased in CS-associated
breast cancers compared with sporadic cases.47 48 Women with CS also have an increased risk (67%)
of benign breast disease characterised by mammary
hamartomas that can be multiple and bilateral.49
Similar to TP53 mutation carriers, PTEN mutation
carriers are advised to have breast surveillance and
interventions as recommended for BRCA mutation
carriers. The testing criteria for CS are those who
present with breast cancer, endometrial cancer,
follicular thyroid cancer, multiple gastrointestinal
hamartomas, ganglioneuromas, or other diseases
including macrocephaly, macular pigmentation of
glans penis, and mucocutaneous lesions.19
Moderate- and low-penetrance genes
PALB2
PALB2 (partner and localiser of BRCA2) is involved
in homologous recombination and double-strand
break repair along with BRCA2.50 51 Loss-of-function mutations are associated with a 2 to 4 times higher
risk than non-mutation carriers for familial breast
cancer.52 53 54 A study analysed the risk of breast
cancer among 362 members of 154 families who had
deleterious PALB2 mutations.55 The results revealed
that the risk of having breast cancer for female
PALB2 mutation carriers was 8 to 9 times higher
among those younger than 40 years, 6 to 8 times higher
among those 40 to 60 years, and 5 times higher
among those >60 years when compared with the
general population. The estimated cumulative risk
of breast cancer among female mutation carriers
increased from 14% to 35% from the age of 50 to 70
years. In addition, the risk of breast cancer for PALB2
mutation carriers was significantly increased by
familial factor.56 Thus, it has been advised that PALB2
mutation testing should be performed routinely to
identify mutations in HBOC families since it may
be of clinical relevance. This is increasingly being
tested.
Other hereditary breast cancer susceptibility genes
There are other low-penetrance genes that are
associated with hereditary breast cancer such
as STK11, CDH1, and MMR genes, and that are
responsible for Peutz-Jeghers syndrome, hereditary
diffuse gastric cancer syndrome, and Lynch
syndrome, respectively.57 58 59 Some moderate-penetrance
genes such as CHEK2, ATM, BRIP1, RAD51C, RAD51D, BARD1, MRE11, RAD50, NBS1, and FANCM have been recognised as breast cancer
susceptibility genes.60
The recent development of multi-gene testing
for hereditary cancer has had a great impact on the
clinical management and genetic counselling of
high-risk patients and their families. The decision
to use multi-gene testing should be no different
than the rationale for testing a single gene. Multi-gene
testing is more cost-effective than sequentially
testing multiple genes associated with a phenotype.
For example, young women diagnosed with breast
cancer can be tested for mutations in BRCA1,
BRCA2, and TP53. Detailed testing criteria for genes
can be found in NCCN guidelines version 1.2016.19
Next-generation sequencing enables simultaneous
analysis of a specific panel of genes, but there are
limited outcome data on clinical interventions,
particularly in lower-penetrance-gene-mutation–related breast cancers. Results of a multi-gene panel
may pose difficulty in interpretation and clinical
decisions. At present, multi-gene testing is largely
performed for research purposes. There are limited
data regarding the degree of cancer risk associated
with some of the genes on the recurrent multi-gene
test. There is a lack of well-established guidelines for
risk management for carriers of mutations in some
of the genes, which may lead to extra surveillance
and surgeries.
Nonetheless multi-gene testing is more cost-effective
and time-effective than single-gene testing,
and provides a higher mutation detection rate. It
may reduce the number of high-risk families with
negative results of finding a gene mutation due to the
increased coverage. The lifetime breast cancer risk
estimates associated with gene mutations are listed
in Table 2.10 13 36 56 58 61 62 63 64 65 66 67 68 69 70 71 72 73 74
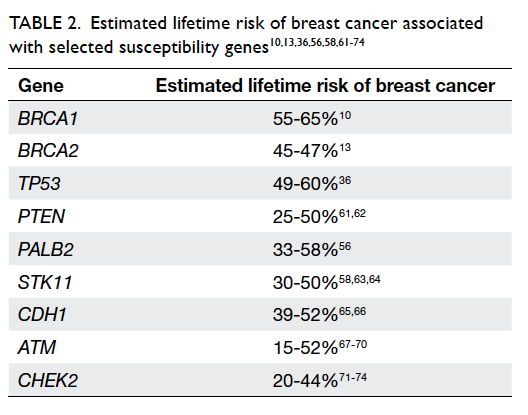
Table 2. Estimated lifetime risk of breast cancer associated with selected susceptibility genes10 13 36 56 58 61 62 63 64 65 66 67 68 69 70 71 72 73 74
In Hong Kong, breast cancer is the most
common cancer in the female population. The Hong
Kong Hereditary Breast Cancer Family Registry
was established in 2007. It functions as a data
registry of hereditary breast, ovarian and prostate
cancer families and is also an established charitable
organisation that subsidises the cost of genetic
testing for underprivileged individuals. More than
1900 patients with breast and/or ovarian cancer
who satisfied the selection criteria have received
genetic testing in Hong Kong. Each individual
underwent thorough genetic counselling to ensure
the implications of genetic testing were understood.
Around 600 probands were screened for BRCA1
and BRCA2 mutations by bi-directional Sanger
sequencing of all coding exons and multiplex ligation-dependent
probe amplification.75 The sensitivity of
identifying mutations is comparable with the gold-standard
method with good bioinformatics support.
Next-generation sequencing meets rigorous quality
standards and can provide clinical sequencing results
that are equivalent to those obtained from Sanger
DNA sequencing analysis.76 We started employing
next-generation DNA sequencing to expedite
analysis workflow and expand the gene panel in 2011
to include TP53 and PTEN for sequencing. Cases
with a negative result after screening with our in-house
developed gene panel are further sequenced
using 454 GS Junior System (Roche Life Sciences) or MiSeq (Illumina). Sequencing
data are analysed by an in-house fully developed
automatic bioinformatics pipeline. The mutation
screening result of a 4-gene panel BRCA1, BRCA2,
TP53, and PTEN in our recruited patients revealed
that 9% carried such mutations. Nonetheless a
number of clinically high-risk patients have tested
negative for the above genes. This indicates that
there is further potential in expanding the coverage
to different lower-penetrance genes such as PALB2,
which has recently been reported to be important
to cause hereditary breast cancer in our testing
strategy.56
Conclusions
Clinical assessment of an individual’s risk of
hereditary cancer is based on the evaluation of family
history, age of onset, and type of cancer. Advances in
molecular genetics testing have identified a number
of genes associated with inherited susceptibility
to breast and/or ovarian cancers such as BRCA1,
BRCA2, PTEN, and TP53. The recent introduction of
next-generation sequencing technology and multi-gene
panel testing for hereditary cancer has rapidly
altered the clinical approach to high-risk patients
and their families. Although there are still limitations,
individuals with hereditary or familial breast/ovarian
cancer are likely to benefit from strategies including
prevention, screening, and targeted treatment.
Suitable patients and families should be offered
genetic counselling and testing.
Acknowledgements
This study was supported by The Hong Kong Hereditary Breast
Cancer Family Registry, Hong Kong Sanatorium and
Hospital, Dr. Ellen Li Charitable Foundation, the
Kerry Group Kuok Foundation Limited and Health
and Medical Research Fund (1123176).
References
1. GLOBOCAN 2012: Estimated cancer incidence, mortality
and prevalence worldwide in 2012. Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed
Jan 2016.
2. Carroll JC, Cremin C, Allanson J, et al. Hereditary breast
and ovarian cancers. Cab Fam Physician 2008;54:1691-2.
3. Daly MB, Axilbund JE, Buys S, et al. Genetic/familial high-risk
assessment: breast and ovarian. J Natl Compr Canc
Netw 2010;8:562-94.
4. Claus EB, Schildkraut JM, Thompson WD, Risch NJ. The
genetic attributable risk of breast and ovarian cancer.
Cancer 1996;77:2318-24. Crossref
5. Pharoah PD, Antoniou A, Bobrow M, Zimmern RL, Easton
DF, Ponder BA. Polygenic susceptibility to breast cancer
and implications for prevention. Nat Genet 2002;31:33-6. Crossref
6. Whittemore AS, Gong G, Itnyre J. Prevalence and
contribution of BRCA1 mutations in breast cancer and
ovarian cancer: results from three U.S. population-based
case-control studies of ovarian cancer. Am J Hum Genet
1997;60:496-504.
7. Peng S, Lü B, Ruan W, Zhu Y, Sheng H, Lai M. Genetic
polymorphisms and breast cancer risk: evidence from
meta-analyses, pooled analyses, and genome-wide
association studies. Breast Cancer Res Treat 2011;127:309-24. Crossref
8. Hall JM, Lee MK, Newman B, et al. Linkage of early-onset
familial breast cancer to chromosome 17q21. Science
1990;250:1684-9. Crossref
9. Wooster R, Neuhausen SL, Mangion J, et al. Localization of
a breast cancer susceptibility gene, BRCA2, to chromosome
13q12-13. Science 1994;265:2088-90. Crossref
10. Chen S, Parmigiani G. Meta-analysis of BRCA1 and
BRCA2 penetrance. J Clin Oncol 2007;25:1329-33. Crossref
11. Mavaddat N, Peock S, Frost D, et al. Cancer risks for BRCA1
and BRCA2 mutation carriers: results from prospective
analysis of EMBRACE. J Natl Cancer Inst 2013;105:812-22. Crossref
12. Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity
and penetrance analysis of the BRCA1 and BRCA2 genes
in breast cancer families. The Breast Cancer Linkage
Consortium. Am J Human Genet 1998;62:676-89. Crossref
13. Antoniou A, Pharoah PD, Narod S, et al. Average risks
of breast and ovarian cancer associated with BRCA1 or
BRCA2 mutations detected in case series unselected for
family history: a combined analysis of 22 studies. Am J
Hum Genet 2003;72:1117-30. Crossref
14. King MC, Marks JH, Mandell JB; New York Breast Cancer
Study Group. Breast and ovarian cancer risks due to
inherited mutations in BRCA1 and BRCA2. Science
2003;302:643-6. Crossref
15. Graeser MK, Engel C, Rhiem K, et al. Contralateral breast
cancer risk in BRCA1 and BRCA2 mutation carriers. J Clin
Oncol 2009;27:5887-92. Crossref
16. Atchley DP, Albarracin CT, Lopez A, et al. Clinical
and pathologic characteristics of patients with BRCA-positive
and BRCA-negative breast cancer. J Clin Oncol
2008;26:4282-8. Crossref
17. Lakhani SR, Reis-Filho JS, Fulford L, et al. Prediction
of BRCA1 status in patients with breast cancer using
estrogen receptor and basal phenotype. Clin Cancer Res
2005;11:5175-80. Crossref
18. Eerola H, Heikkilä P, Tamminen A, Aittomäki K, Blomqvist
C, Nevanlinna H. Histopathological features of breast
tumours in BRCA1, BRCA2 and mutation-negative breast
cancer families. Breast Cancer Res 2005;7:R93-100. Crossref
19. Genetic/Familial High-Risk Assessment: Breast and
Ovarian 2016. Available from: http://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed Mar 2016.
20. Susman E. Metastatic TNBC: No improvement with
carboplatin compared with docetaxel—study did,
however, show importance of molecular testing to
identify responsive subsets. Oncology Times 2015;37:42-3. Crossref
21. Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose)
polymerase inhibitor olaparib in patients with BRCA1 or
BRCA2 mutations and advanced breast cancer: a proof-of-concept
trial. Lancet 2010;376:235-44. Crossref
22. Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose)
polymerase inhibitor olaparib in patients with
BRCA1 or BRCA2 mutations and recurrent ovarian
cancer: a proof-of-concept trial. Lancet 2010;376:245-51. Crossref
23. Diller L, Kassel J, Nelson CE, et al. p53 functions as a cell
cycle control protein in osteosarcomas. Mol Cell Biol
1990;10:5772-81. Crossref
24. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe
SW. Oncogenic ras provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell
1997;88:593-602. Crossref
25. Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J.
Induction of apoptosis by wild-type p53 in a human
colon tumor-derived cell line. Proc Natl Acad Sci U S A 1992;89:4495-9. Crossref
26. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev
Cancer 2009;9:691-700. Crossref
27. Wang PY, Ma W, Park JY, et al. Increased oxidative
metabolism in the Li-Fraumeni syndrome. N Engl J Med
2013;368:1027-32. Crossref
28. Wang Y, Blandino G, Oren M, Givol D. Induced p53
expression in lung cancer cell line promotes cell senescence
and differentially modifies the cytotoxicity of anti-cancer
drugs. Oncogene 1998;17:1923-30. Crossref
29. Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer,
and other neoplasms. A familial syndrome? Ann Intern
Med 1969;71:747-52. Crossref
30. Li FP, Fraumeni JF Jr, Mulvihill JJ, et al. A cancer
family syndrome in twenty-four kindreds. Cancer Res
1988;48:5358-62.
31. McCuaig JM, Armel SR, Novokmet A, et al. Routine TP53
testing for breast cancer under age 30: ready for prime
time? Fam Cancer 2012;11:607-13. Crossref
32. Sidransky D, Tokino T, Helzlsouer K, et al. Inherited p53
gene mutations in breast cancer. Cancer Res 1992;52:2984-6.
33. Birch JM, Blair V, Kelsey AM, et al. Cancer phenotype
correlates with constitutional TP53 genotype in families
with the Li-Fraumeni syndrome. Oncogene 1998;17:1061-8. Crossref
34. Olivier M, Goldgar DE, Sodha N, et al. Li-Fraumeni and
related syndromes: correlation between tumor type, family
structure, and TP53 genotype. Cancer Res 2003;63:6643-50.
35. Walsh T, Casadei S, Coats KH, et al. Spectrum of mutations
in BRCA1, BRCA2, CHEK2, and TP53 in families at high
risk of breast cancer. JAMA 2006;295:1379-88. Crossref
36. Masciari S, Dillon DA, Rath M, et al. Breast cancer
phenotype in women with TP53 germline mutations: a Li-Fraumeni syndrome consortium effort. Breast Cancer Res Treat 2012;133:1125-30. Crossref
37. Melhem-Bertrandt A, Bojadzieva J, Ready KJ, et al. Early
onset HER2-positive breast cancer is associated with
germline TP53 mutations. Cancer 2012;118:908-13. Crossref
38. Wilson JR, Bateman AC, Hanson H, et al. A novel HER2-positive breast cancer phenotype arising from germline
TP53 mutations. J Med Genet 2010;47:771-4. Crossref
39. Lalloo F, Varley J, Moran A, et al. BRCA1, BRCA2 and
TP53 mutations in very early-onset breast cancer with
associated risks to relatives. Eur J Cancer 2006;42:1143-50. Crossref
40. Mouchawar J, Korch C, Byers T, et al. Population-based
estimate of the contribution of TP53 mutations to
subgroups of early-onset breast cancer: Australian Breast
Cancer Family Study. Cancer Res 2010;70:4795-800. Crossref
41. Varley JM. Germline TP53 mutations and Li-Fraumeni
syndrome. Hum Mutat 2003;21:313-20. Crossref
42. Birch JM, Hartley AL, Tricker KJ, et al. Prevalence and
diversity of constitutional mutations in the p53 gene
among 21 Li-Fraumeni families. Cancer Res 1994;54:1298-304.
43. Shen WH, Balajee AS, Wang J, et al. Essential role for
nuclear PTEN in maintaining chromosomal integrity. Cell
2007;128:157-70. Crossref
44. Hanssen AM, Fryns JP. Cowden syndrome. J Med Genet
1995;32:117-9. Crossref
45. Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng
C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012;18:400-7. Crossref
46. Pilarski R. Cowden syndrome: a critical review of the
clinical literature. J Genet Couns 2009;18:13-27. Crossref
47. Schrager CA, Schneider D, Gruener AC, Tsou HC,
Peacocke M. Similarities of cutaneous and breast pathology
in Cowden’s Syndrome. Exp Dermatol 1998;7:380-90. Crossref
48. Schrager CA, Schneider D, Gruener AC, Tsou HC,
Peacocke M. Clinical and pathological features of breast
disease in Cowden’s syndrome: an underrecognized
syndrome with an increased risk of breast cancer. Hum
Pathol 1998;29:47-53. Crossref
49. Starink TM, van der Veen JP, Arwert F, et al. The Cowden
syndrome: a clinical and genetic study in 21 patients. Clin
Genet 1986;29:222-33. Crossref
50. Xia B, Sheng Q, Nakanishi K, et al. Control of BRCA2
cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 2006;22:719-29. Crossref
51. Sy SM, Huen MS, Zhu Y, Chen J. PALB2 regulates
recombinational repair through chromatin association and
oligomerization. J Biol Chem 2009;284:18302-10. Crossref
52. Tischkowitz M, Xia B. PALB2/FANCN: recombining
cancer and Fanconi anemia. Cancer Res 2010;70:7353-9. Crossref
53. Casadei S, Norquist BM, Walsh T, et al. Contribution of
inherited mutations in the BRCA2-interacting protein
PALB2 to familial breast cancer. Cancer Res 2011;71:2222-9. Crossref
54. Erkko H, Xia B, Nikkilä J, et al. A recurrent mutation in
PALB2 in Finnish cancer families. Nature 2007;446:316-9. Crossref
55. Lee AS, Ang P. Breast-cancer risk in families with mutations
in PALB2. N Engl J Med 2014;371:1650-1. Crossref
56. Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer
risk in families with mutations in PALB2. N Engl J Med
2014;371:497-506. Crossref
57. Fitzgerald RC, Hardwick R, Huntsman D, et al. Hereditary
diffuse gastric cancer: updated consensus guidelines for
clinical management and directions for future research. J
Med Genet 2010;47:436-44. Crossref
58. Hearle N, Schumacher V, Menko FH, et al. Frequency and
spectrum of cancers in the Peutz-Jeghers syndrome. Clin
Cancer Res 2006;12:3209-15. Crossref
59. Win AK, Young JP, Lindor NM, et al. Colorectal and other
cancer risks for carriers and noncarriers from families
with a DNA mismatch repair gene mutation: a prospective
cohort study. J Clin Oncol 2012;30:958-64. Crossref
60. Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA:
new hereditary breast cancer susceptibility genes. Cancer
Treat Rev 2015;41:1-8. Crossref
61. Bennett KL, Mester J, Eng C. Germline epigenetic
regulation of KILLIN in Cowden and Cowden-like
syndrome. JAMA 2010;304:2724-31. Crossref
62. Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an
overview. Genet Med 2009;11:687-94. Crossref
63. Lim W, Hearle N, Shah B, et al. Further observations on
LKB1/STK11 status and cancer risk in Peutz-Jeghers
syndrome. Br J Cancer 2003;89:308-13. Crossref
64. McGarrity TJ, Amos CI, Frazier ML, Wei C. Peutz-Jeghers
syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al,
editors. GeneReviews [internet]. Seattle (WA): GeneReviews(R); 1993.
65. Pharoah PD, Guilford P, Caldas C; International Gastric
Cancer Linkage Consortium. Incidence of gastric cancer
and breast cancer in CDH1 (E-cadherin) mutation
carriers from hereditary diffuse gastric cancer families.
Gastroenterology 2001;121:1348-53. Crossref
66. Kaurah P, MacMillan A, Boyd N, et al. Founder and
recurrent CDH1 mutations in families with hereditary
diffuse gastric cancer. JAMA 2007;297:2360-72. Crossref
67. Renwick A, Thompson D, Seal S, et al. ATM mutations that
cause ataxia-telangiectasia are breast cancer susceptibility
alleles. Nat Genet 2006;38:873-5. Crossref
68. Thompson D, Duedal S, Kirner J, et al. Cancer risks and
mortality in heterozygous ATM mutation carriers. J Natl
Cancer Inst 2005;97:813-22. Crossref
69. Ahmed M, Rahman N. ATM and breast cancer
susceptibility. Oncogene 2006;25:5906-11. Crossref
70. Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients
with hereditary pancreatic cancer. Cancer Discov
2012;2:41-6. Crossref
71. Meijers-Heijboer H, van den Ouweland A, Klijn J, et al.
Low-penetrance susceptibility to breast cancer due to
CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2
mutations. Nat Genet 2002;31:55-9. Crossref
72. Narod SA. Testing for CHEK2 in the cancer genetics clinic:
ready for prime time? Clin Genet 2010;78:1-7. Crossref
73. Weischer M, Bojesen SE, Ellervik C, Tybjaerg-Hansen
A, Nordestgaard BG. CHEK2*1100delC genotyping for
clinical assessment of breast cancer risk: meta-analyses
of 26,000 patient cases and 27,000 controls. J Clin Oncol
2008;26:542-8. Crossref
74. Cybulski C, Wokołorczyk D, Jakubowska A, et al. Risk
of breast cancer in women with a CHEK2 mutation with
and without a family history of breast cancer. J Clin Oncol
2011;29:3747-52. Crossref
75. Kwong A, Chen J, Shin VY, et al. The importance of analysis
of long-range rearrangement of BRCA1 and BRCA2 in
genetic diagnosis of familial breast cancer. Cancer Genet
2015;208:448-54. Crossref
76. McCourt CM, McArt DG, Mills K, et al. Validation of
next generation sequencing technologies in comparison
to current diagnostic gold standards for BRAF, EGFR and
KRAS mutational analysis. PloS One 2013;8:e69604. Crossref