DOI: 10.12809/hkmj134085
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Novel use of idebenone in Leber’s hereditary optic neuropathy in Hong Kong
SW Cheng, MB, ChB, MRCPCH1; CH Ko, FHKAM (Paediatrics), FRCP RCPS (Glasg)1; SK Yau, MB, ChB, FRCSEd2; Chloe Mak, MD, PhD3; YF Yuen, FRCSEd, FHKAM (Ophthalmology)2; CY Lee, FHKAM (Paediatrics), FRCP (Edin)1
1 Department of Paediatrics and Adolescent Medicine, Caritas Medical
Centre, Shamshuipo, Hong Kong
2 Department of Ophthalmology, Caritas Medical Centre, Shamshuipo,
Hong Kong
3 Department of Pathology, Princess Margaret Hospital, Laichikok, Hong
Kong
Corresponding author: Dr SW Cheng (csw350@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
We report a case of a young Chinese male presenting
with sequential, painless, bilateral visual loss in Hong
Kong. He was diagnosed to have Leber’s hereditary
optic neuropathy with genetic workup showing
G11778A mutation with over 80% heteroplasmy.
He was started on idebenone treatment 11 months
after onset of the binocular disease. To our best
knowledge, this is the first case of Leber’s hereditary
optic neuropathy treated with idebenone in Hong
Kong. The recent evidence of the diagnosis and
treatment of this devastating disease is reviewed.
Introduction
Leber’s hereditary optic neuropathy (LHON) is the
commonest mitochondrial disease found to affect
around 1 in 30 000 people in the first population-based
clinical and molecular genetic study carried
out in the UK.1 It is characterised by sequential
bilateral visual loss, optic nerve dysfunction, and
retinal ganglion cell degeneration. Characteristic
visual field defect in LHON includes either central or
cecocentral scotomas. Approximately 70% of patients
were found to be young adults in the UK study. Over
20 mitochondrial DNA point mutations have been
reported in LHON worldwide.2 Around 95% of the
LHON pedigree have one of the three mitochondrial
DNA point mutations namely, G3460A, G11778A,
and T14484C, which are responsible for coding of
complex I subunits of the mitochondrial respiratory
chain.
Case report
A 15-year-old Chinese male, with a history of
congenital red-green colour blindness, presented
with sudden blurring of right eye central vision in
August 2011. He did not experience any ocular pain
or photopsia. There was no family history of ocular
or neurological diseases.
On admission, his visual acuity was 6/120
in the right eye and 6/6 in the left eye. Right-eye
visual field examination showed central scotoma
associated with right relative afferent papillary
defect. Fundoscopy revealed right eye disc swelling
without peripapillary haemorrhage or exudate (Fig a). Neurological and cardiovascular examinations
were normal.
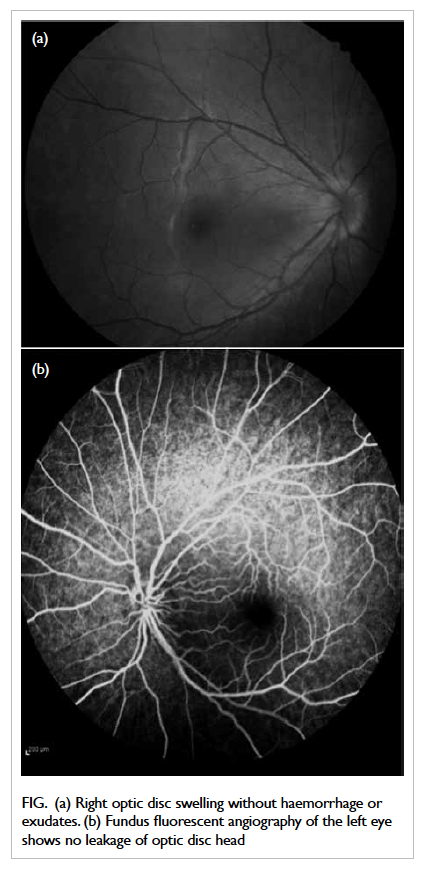
Figure. (a) Right optic disc swelling without haemorrhage or exudates. (b) Fundus fluorescent angiography of the left eye shows no leakage of optic disc head
Perimetry revealed right centrocecal scotoma.
Computed tomography of brain and orbit with
contrast showed normal eye globes and extra-ocular
muscles but mildly thickened intra-orbital right optic
nerve suggestive of right optic neuritis. Erythrocyte
sedimentation rate and C-reactive protein were not
elevated. Blood test for anti-nuclear antibody was
negative. Serologies for toxoplasmosis, herpes virus,
Lyme disease and syphilis as well as tuberculin test
were negative. Visual evoked potential revealed
prolonged P100 over the right eye compatible with
optic neuropathy. Magnetic resonance imaging of the
brain and orbit was unremarkable. The patient was
diagnosed to have optic neuritis and recommended
for steroid treatment according to Optic Neuritis
Treatment Trial (ONTT).3 However, his parents
declined the treatment due to the possible side-effects
of steroids.
Two months later, the patient experienced
sudden painless blurring of left eye central vision.
Visual acuity was 6/60 in the left eye while it
remained at 6/120 in the right eye. Cerebrospinal
fluid examination revealed normal biochemistry and
cell count, and was negative for oligoclonal bands.
Serum folate and vitamin B12 levels were normal.
He was started on an ONTT-recommended dose of
intravenous methylprednisolone (1000 mg/day) for
3 days, followed by oral prednisolone (1 mg/kg/day)
for 11 days. However, his visual function deteriorated
despite treatment. In view of the atypical features
of sequential visual bilateral optic neuritis without
recovery, LHON was suspected and confirmed
by identification of mitochondrial DNA point
mutation at 11778 guanine to adenine in the blood,
with high mutant load heteroplasmy (over 80%).
Fundus fluorescent angiography revealed left-disc
telangiectatic vessels without optic disc head leakage
compatible with typical angiographic features of
LHON (Fig b).
He was then put on high-dose oral antioxidants
(vitamin C 500 mg daily and co-enzyme Q10
ubiquinone). The medication was switched to
idebenone 900 mg per day in divided doses when the
drug became available in the hospital.
Six months after treatment with idebenone,
his visual acuity remained static: 1/60 and 2/60 in
the right and left eye, respectively. There were no
side-effects related to the treatment. He gradually
adapted to school life, with improvement in using
Braille. Genetic study revealed the mother to be a
LHON carrier (G11778A). His sister refused genetic
testing due to the possible implications on future
employment and academic considerations.
Discussion
Optic neuritis is an inflammatory disease of the optic
nerve. It is the second commonest acquired optic
nerve disorder in persons under 50 years old. The typical
presenting symptom includes sudden visual loss
with dyschromatopsia over several hours to days.
This may be associated with dull retrobulbar pain
which worsens with ocular movement. Symptoms
can occur in both eyes either simultaneously or
sequentially. A clinical diagnosis of optic neuritis may
be made based on the following signs and symptoms:
sudden visual loss with progression within 1 week,
dyschromatopsia, pain on extra-ocular movement,
no features of uveitis, and vision improvement
beginning in 3 to 4 weeks.4 According to the findings
in ONTT, 80% of patients with optic neuritis will
improve within the first 3 weeks.5 Over 95% will
regain visual acuity of at least 20/40, regardless of
treatment options at 12 months’ time from onset of
symptoms.6 The initial presentation of our patient
was similar to that of optic neuritis. However, failure
of spontaneous recovery in the absence of other
causes should alert the clinician to consider atypical
optic neuritis. The differential diagnoses of atypical
optic neuritis are listed in the Table. A detailed
history and clinical examination helps to understand
the underlying aetiology, which may be confirmed
by relevant radiological, serological, bacteriological,
electrophysiological, and molecular investigations.
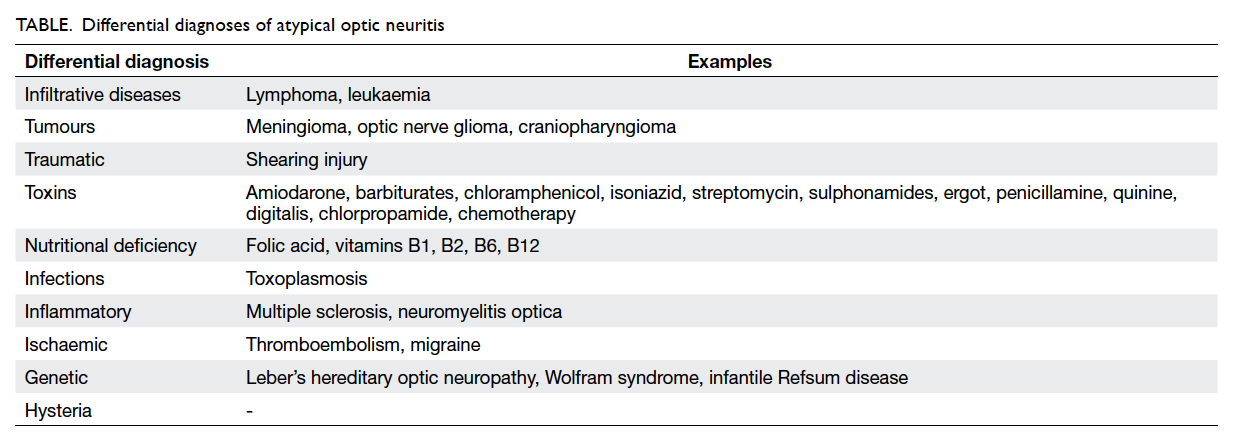
Table. Differential diagnoses of atypical optic neuritis
Patients with LHON experience devastating
visual loss, typically worse than 20/200 in both eyes.
At least 97% of patients will have sequential eye
involvement within 1 year.7 Over 90% of patients
harbour one of the three pathogenic mitochondrial
mutations (G3460A, G11778A, T14484C), which
affect complex I of the mitochondrial respiratory
chain.8 The pathogenesis of LHON involves a
combination of decreased complex I–driven
adenosine triphosphate production, increase in free
radical production and, finally, retinal ganglion cell
apoptosis.9 The most important prognostic factor for
visual recovery is the mutation status. The T14484C
mutation carries the best chance of some degree of
visual improvement in 37% to 71% of patients, while
the G11778A mutation is associated with only 4%
chance of recovery.7
Kirkman et al10 suggested that visual loss
occurs more often in smokers and people with high
alcohol intake. It is essential to advise patients to
avoid tobacco smoking, excessive alcohol intake,
and medications that may have mitochondrial
toxicity (eg aminoglycosides, metformin, statins),
especially during the acute phase of visual loss.
Directed therapies for mitochondrial disorders are
very limited. To date, there is no effective treatment
for LHON. The mainstay of management remains
supportive, such as low visual aids to assist reading,
communication, and employment.
Anecdotal reports have shown beneficial effects
of idebenone, which is a short-chain benzoquinone
structurally related to co-enzyme Q10. It is a potent
antioxidant and inhibitor of lipid peroxidation. It
facilitates mitochondrial electron flux in bypassing
complex I.11 In a retrospective open-label study
involving 28 LHON patients, Mashima et al12
reported significantly shortened onset of visual
recovery (11.1 months vs 17.4 months; P=0.03) and
shortened interval for recovery of vision to ≥0.3 (17.6
months vs 34.4 months; P=0.01) in the treated group
versus the untreated group. Jancic13 administered
idebenone to nine patients at 135 mg/day for
up to 1 year. Three patients reported subjective
improvement in visual acuity, one with monocular
disease showed arrest of progression of visual loss to
the other eye, and four demonstrated improvement
in visual evoked potential. Klopstock et al14
conducted a 24-week multicentre double-blind,
randomised, placebo-controlled trial in 85 patients
given idebenone at a dose of 900 mg/day. There was
no significant difference in the overall group for best
recovery of visual acuity. However, in the subgroup
with discordant visual acuities at baseline (n=30), a
significant trend for improvement in visual acuity
was observed in the treated group. Idebenone was
well tolerated with no adverse effect. In summary,
current evidence suggests that idebenone is probably
beneficial in the early-stage LHON patients with
discordant visual acuities, and may help to shorten
the interval of visual recovery.
In our patient, the static eye condition in the
initial 6 weeks had prompted extensive diagnostic
investigations including molecular study. However,
the patient developed binocular involvement 2
months after onset of the symptoms. As idebenone
was not available with the local pharmaceutical
company, ordering of the drug from overseas had
involved time and administrative process. The poor
response of visual symptoms to idebenone might be
explained by the late introduction of the treatment,
in this case, 11 months after binocular involvement.
In our patient, the interval from disease onset
till binocular involvement was only 2 months.
Heightened physician awareness is important to
identify this devastating condition during this
interval, which may represent the golden window
for initiation of idebenone to prevent sequential
visual loss and hasten recovery. Molecular analysis
for the three common mutations, which is available
locally, aids definitive diagnosis, prognostication,
and detection of presymptomatic family members.
Those presymptomatic family members with
positive mutation require lifelong surveillance,
lifestyle modification, and prompt intervention at
disease onset.
Acknowledgement
The authors would like to thank Ms Josephine
Lui, pharmacist of Caritas Medical Centre, for her
arduous effort to source idebenone for this patient.
References
1. Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM,
Chinnery PF. The epidemiology of Leber hereditary optic
neuropathy in the North East of England. Am J Hum Genet
2003;72:333-9. CrossRef
2. Brown MD, Wallace DC. Spectrum of mitochondrial DNA
mutations in Leber’s hereditary optic neuropathy. Clin
Neurosci 1994;2:138-45.
3. Beck RW, Cleary PA, Anderson MM Jr, et al. A randomized,
controlled trial of corticosteroids in the treatment of acute
optic neuritis. The Optic Neuritis Study Group. N Engl J
Med 1992;326:581-8. CrossRef
4. Gal RL, Vedula SS, Beck R. Corticosteroids for treating optic
neuritis. Cochrane Database Syst Rev 2012;(4):CD001430.
5. Beck RW, Cleary PA, Backlund JC. The course of visual
recovery after optic neuritis. Experience of the Optic
Neuritis Treatment Trial. Ophthalmology 1994;101:1771-8. CrossRef
6. Beck RW, Cleary PA. Optic neuritis treatment trial. One-year
follow-up results. Arch Ophthalmol 1993;111:773-5. CrossRef
7. Newman NJ, Biousse V, David R, et al. Prophylaxis
for second eye involvement in Leber hereditary optic
neuropathy: an open-labeled, nonrandomized multicenter
trial of topical brimonidine purite. Am J Ophthalmol
2005;140:407-15. CrossRef
8. Harding AE, Sweeney MG, Govan GG, Riordan-Eva P.
Pedigree analysis in Leber hereditary optic neuropathy
families with a pathogenic mtDNA mutation. Am J Hum
Genet 1995;57:77-86.
9. Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology
of mitochondrial disease. Surv Ophthalmol
2010;55:299-334. CrossRef
10. Kirkman MA, Yu-Wai-Man P, Korsten A, et al. Gene-environment
interactions in Leber hereditary optic
neuropathy. Brain 2009;132:2317-26. CrossRef
11. Haefli RH, Erb M, Gemperli AC, et al. NQO1-dependent
redox cycling of idebenone: effects on cellular redox
potential and energy levels. PLoS One 2011;6:e17963. CrossRef
12. Mashima Y, Kigasawa K, Wakakura M, Oguchi Y. Do
idebenone and vitamin therapy shorten the time to achieve
visual recovery in Leber hereditary optic neuropathy? J
Neuroophthalmol 2000;20:166-70. CrossRef
13. Jancic J. Effectiveness of idebenone therapy in Leber’s
hereditary optic neuropathy. Proceedings of 11th
European College of Neuropsychopharmacology (ECNP)
Regional Meeting; 2011 Apr 14-16; St. Petersburg, Russian
Federation. Amsterdam: Elsevier; 2011; 21: S175.
14. Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A
randomized placebo-controlled trial of idebenone in
Leber’s hereditary optic neuropathy. Brain 2011;134:2677-86. CrossRef