DOI: 10.12809/hkmj134041
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Perforin gene mutation in familial haemophagocytic lymphohistiocytosis: the first reported case from Hong Kong
Grace PK Chiang, MB, ChB, MRCPCH1; CK Li, MD, FHKAM (Paediatrics)1; Vincent Lee, MB, BS, FHKAM (Paediatrics)1; Frankie WT Cheng, MD, FHKAM (Paediatrics)1; Alex WK Leung, MB, ChB, FHKAM (Paediatrics)1; Shinsaku Imashuku, MD2; Toshihiko Imamura, MD, PhD3; Matthew MK Shing, MB, BS, FHKAM (Paediatrics)1
1 Department of Paediatrics, Prince of Wales Hospital, The Chinese
University of Hong Kong, Shatin, Hong Kong
2 Division of Pediatrics and Hematology, Takasago-seibu Hospital,
Takasago, Japan
3 Department of Pediatrics, Kyoto, Prefectural University of Medicine,
Graduate School of Medical Science, Japan
Corresponding author: Dr Grace PK Chiang (gpkchiang@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Familial haemophagocytic lymphohistiocytosis is a
rare but invariably fatal disease without haematopoietic
stem cell transplantation. Genetic defect identification
is useful for confirming a clinical diagnosis,
predicting the risk of future recurrence, and defining
haemophagocytic lymphohistiocytosis predisposition
in asymptomatic family members. Notably, familial haemophagocytic lymphohistiocytosis type 2
associates with mutations in the perforin gene (PRF1) which is the most frequent subtype of familial
haemophagocytic lymphohistiocytosis. Although
perforin gene mutations have been described in
Asians, they are largely reported from Japan. The case
reported here is the first familial haemophagocytic lymphohistiocytosis type 2 patient in Hong Kong
with an identified perforin gene mutation.
Introduction
Haemophagocytic lymphohistiocytosis (HLH) is
characterised by fever, hepatosplenomegaly, central
nervous system symptoms, cytopenia, coagulopathy,
and lipid changes because of pathological immune
activation, hypercytokinaemia and organ infiltration
by phagocytosing histiocytes. Despite being an
aggressive disease, effective treatment does exist. A
treatment protocol was firstly designed in 19941 and
later revised in 2004 by the HLH Study Group of the
Histiocyte Society.2 Since the implementation of the
treatment protocol of HLH 1994, its 5-year survival
rate has improved from around 20% to more than
50%.1
Notably, HLH is comprised of two different
conditions: familial/primary HLH (FHL) and
secondary HLH (Table 1),3 with the former being
an autosomal recessive condition. Five mutations
that lead to FHL (Nos 1-5) have now been identified
and the underlying genetic defect described in four.
They are: PRF1, UNC13D, STX11, and STXBP2.
For genetic defect in FHL1, the potential gene
locus has been identified but not the specific
genetic defect. The perforin gene (PRF1) mutation
is the first genetic defect described in FHL (FHL2)
and accounts for 20% to 50% of all affected FHL
families identified in a Japanese study.4 Perforin is a
soluble, pore-forming cytolytic protein synthesised in cytotoxic lymphocytes. This molecule plays a
crucial role in regulating the access of granzymes
to the cytosol of target cells, where they cleave
key substrates to initiate apoptotic cell death. It is
sequestered, along with granzyme serine proteases,
in secretory cytotoxic granules. The PRF1 mutation
results in reduction of perforin protein production
and its cytotoxic function. This in turn impairs the
control of lymphocyte homeostasis during immune
responses and leads to hypercytokinaemia and
continued expansion of populations of histiocytes
and activated cytotoxic lymphocytes.5 Patients
without an identifiable genetic cause but with a clear
familial history of HLH are also classified as having
FHL.
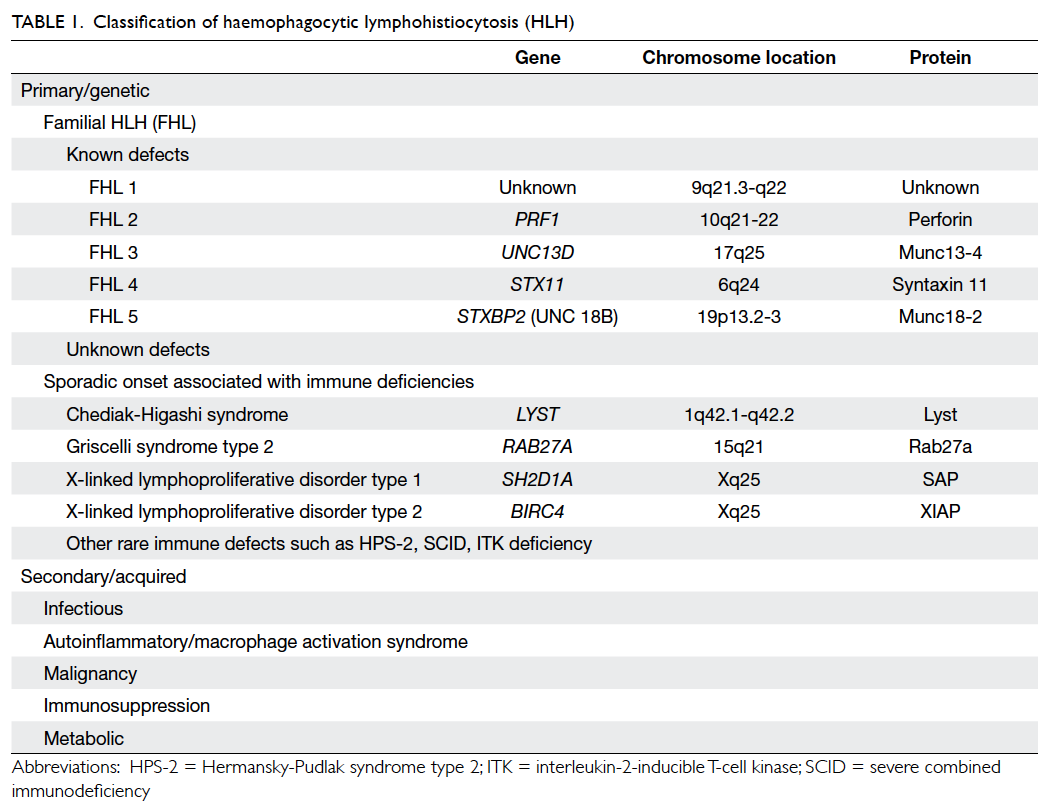
Table 1. Classification of haemophagocytic lymphohistiocytosis (HLH)
The differentiation of primary and secondary
HLH is notoriously difficult. The only definite way
is by genetic study to find the causative mutation.
In Asia, the perforin gene mutation has mostly
been identified in HLH patients in Japan.6 7 The case
reported here is the first and the only HLH perforin
gene mutation identified in Hong Kong.
Case report
The patient was the first child in a non-consanguineous
family, with no history of previous unexplained
deaths in the parents’ families. The child presented
at 34 days of life with fever, hepatosplenomegaly, and pancytopenia in June 2009. The ferritin level was
markedly increased to 18 513 (reference range [RR],
29-333) pmol/L and there was hypofibrinogenaemia
with a level of 0.54 (RR, 1.85-3.83) g/L. Bone marrow
examination showed features of haemophagocytosis.
The diagnostic criteria for HLH were therefore
met. Initial magnetic resonance imaging (MRI) of
the brain and cerebrospinal fluid examination were
normal. Virology investigations including serology
for Epstein-Barr virus (EBV), human herpesvirus 6,
herpes simplex virus, and cytomegalovirus were all negative. The patient was treated according to HLH
2004 protocol with dexamethasone, cyclosporine A,
and etoposide. Her clinical condition deteriorated
with severe metabolic acidosis and she underwent
haemodialysis. She experienced persistent
neutropenia after the first dose of etoposide.
Repeat bone marrow examinations showed
markedly depressed granulopoiesis with residual
haemophagocytic activity. Further doses of etoposide
were therefore withheld while dexamethasone and
cyclosporine A were continued. The first course
of chemotherapy was stopped after the 11th week
of treatment. However, the patient had a relapse
of HLH 3 weeks after stopping chemotherapy
(manifesting as fever and hepatosplenomegaly).
Repeat bone marrow examination confirmed
the presence of haemophagocytic activity, for
which treatment with dexamethasone, etoposide,
and cyclosporine A was restarted. She developed
progressive metabolic acidosis8 that was once
again treated by haemodialysis. Her condition then
became stabilised.
Since this patient presented at early age and
had a recurrence after cessation of chemotherapy,
she was suspected to suffer from FHL, for which
the ultimate treatment is haematopoietic stem cell
transplant (HSCT). Search for a related or unrelated
donor was started while the patient continued to receive chemotherapy.
While waiting for the HSCT, the patient
developed tremors of the lower limbs, and bilateral
ankle clonus, limb spasticity and intermittent
squints (with no definite visual fixation) were
noted. The developmental age regressed from 8
to 3 months, whilst brain MRI revealed diffuse
parenchymal and leptomeningeal enhancing
lesions suggestive of lymphohistiocytic infiltration.
Cerebrospinal fluid also showed presence of
pleocytosis and a lymphohistiocytic infiltrate. The
patient was diagnosed to have central nervous
system involvement by HLH. Three doses of
intrathecal chemotherapy with methotrexate 6 mg
and hydrocortisone 8 mg were given over a 10-day
period.
The patient received an unrelated double-unit cord blood transplant after conditioning with
oral busulphan (23 mg/kg), etoposide (30 mg/kg),
cyclophosphamide (120 mg/kg), and thymoglobulin
(7.5 mg/kg). She had a neutropenic fever on post-transplant
day 12. Donor cell engraftment was
achieved on post-transplant day 16. Regrettably,
she developed veno-occlusive disease causing
hyperbilirubinaemia, fluid retention, progressive
hepatosplenomegaly, and respiratory distress. The
maximum bilirubin level was 209 μmol/L. Despite
intensive care unit treatment, intubation, and
positive pressure ventilation, the patient developed
respiratory failure and died on day 45 after cord blood
transplant. The parents refused a full postmortem. A
postmortem liver biopsy showed marked sinusoidal
dilatation and congestion with atrophy of central
hepatocytes. These features were compatible with sinusoidal obstruction due to veno-occlusive disease.
Genetic analysis of the patient and the parents’
blood was performed, with the coding region of
the perforin gene in exons 2 and 3 amplification
by a polymerase chain reaction. This revealed a
heterozygous one base pair deletion (65 delC) in exon
2 in the patient and her father. There was another
mutation 853-855 del AAG in exon 3 of patient and
her mother. Collectively, the patient had compound
heterozygous mutations of the perforin gene, namely
853-855 del AAG and 65delC.
Discussion
Previously we reported our seven consecutive cases
of HLH encountered from 1991 to 2006.9 Since
then, there had been four other patients. Apart from
showing haemophagocytosis in bone marrow or
lymph node or both (Table 2), all eleven patients had
fever, splenomegaly, and cytopenia in at least two cell
lines. They also had markedly elevated ferritin levels
with or without a raised fasting triglyceride level
or hypofibrinogenaemia. The standard diagnostic
criteria for HLH were met in all patients. Another
teaching hospital in Hong Kong reported nine cases
from 1991 to 2006.10 The overall survival of all 20
patients was 58%.
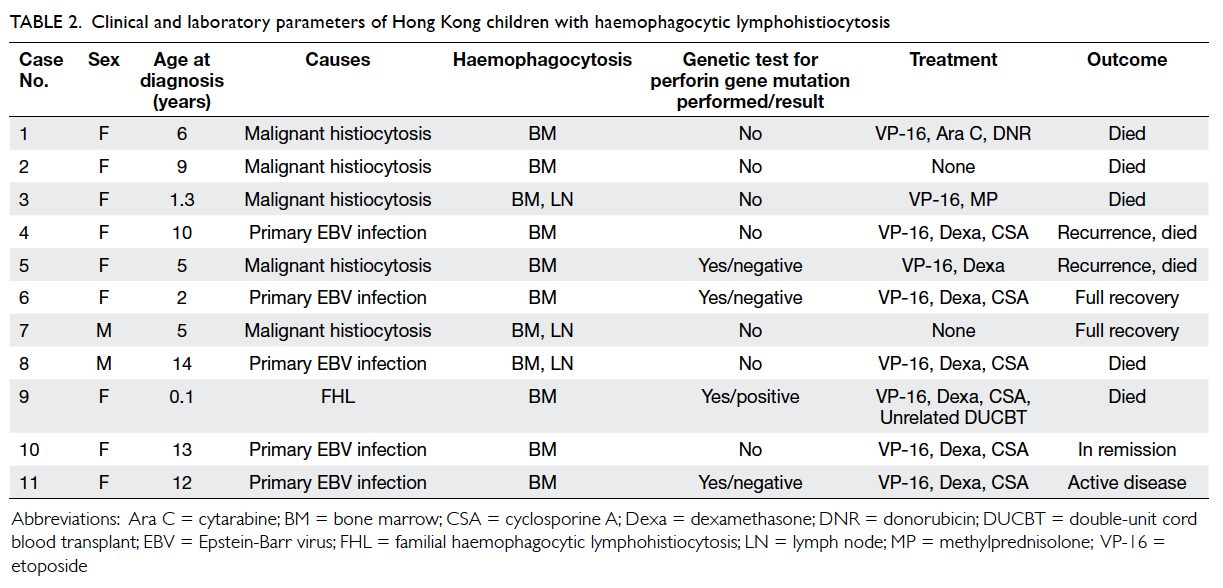
Table 2. Clinical and laboratory parameters of Hong Kong children with haemophagocytic lymphohistiocytosis
Interestingly, EBV infection was confirmed (by
immunoglobulin M vs EBV or EBV DNA) in 11 (55%)
of the 20 patients. The overall survival of EBV-related
HLH was 55%. In eight patients, their secondary HLH
was related to malignant histiocytosis, Still’s disease,
and anaplastic large cell lymphoma. Two patients
had X-linked lymphoproliferative disorders.10 They all had primary EBV infection and one had died. The
other patient received a mismatched unrelated cord
blood transplant and had a full recovery without
recurrence.
Four patients in our unit were investigated
for perforin gene mutations; only one whom had an
abnormality (compound heterozygous mutations
in the gene). Hence this patient is the first reported
case in Hong Kong with a perforin gene mutation
causing FHL.
In Asia, most perforin gene mutations of
HLH patients have been reported from Japan.
Ueda et al6 reported five of 14 HLH patients with
perforin gene abnormalities. The 1090-1091delCT
and 207delC mutations of the perforin gene were
frequently present in Japanese HLH patients. Ueda
et al4 also reported a collaborative study which did
not show PRF1 gene mutations from Korea (n=4),
Malaysia (n=3), Hong Kong (n=2), Australia (n=1),
and Taiwan (n=1). Lee et al11 from Taiwan reported
26 HLH patients; none of whom had PRF1, Mun12-4, STX11, or SH2D1A mutations. There was only
one case report of a heterozygous PRF1 mutation
(Arg390stop) in a young Taiwanese girl who
presented with a panniculitis-like T-cell lymphoma
and subsequently endured fatal HLH.12
Among the 20 patients in Hong Kong, only
one had a PRF1 gene mutation and two had X-linked
lymphoproliferative disorders. However, the
actual rate of genetic abnormalities in HLH patients
remains unknown as not all patients with HLH had
genetic testing. This is partially due to inconsistent
protocols for genetic investigations in this disease
entity and inadequate laboratory support for
genetic tests. Clearly, revision of the local clinical
and laboratory service protocol is warranted, and
more importantly, an international multicentre
collaboration to improve immunological assessment
and genetic analysis of HLH patients should be
promoted.
References
1. Henter JI, Arico M, Egeler RM, et al. HLH-94: a treatment
protocol for hemophagocytic lymphohistiocytosis. HLH
study Group of the Histiocyte Society. Med Pediatr Oncol
1997;28:342-7. CrossRef
2. Henter JI, Home A, Aricó M, et al. HLH-2004: Diagnostic
and therapeutic guidelines for hemophagocytic
lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124-31. CrossRef
3. Freeman HR, Ramanan AV. Review of haemophagocytic
lymphohistiocytosis. Arch Dis Child 2011;96:688-93. CrossRef
4. Ueda I, Ghim T, Peng LH, et al. Proceedings of the 2nd
Congress Asian Society for Pediatric Research; 2006 Dec
8-10; Yokohama, Japan.
5. Usmani GH, Woda BA, Newburger PE. Advances in
understanding the pathogenesis of HLH. Br J Haematol
2013;161:609-22. CrossRef
6. Ueda I, Morimoto A, Inaba T, et al. Characteristic perforin
gene mutations of haemophagocytic lymphohistiocytosis
patients in Japan. Br J Haematol 2003;121:503-10. CrossRef
7. Ueda I, Kurokawa Y, Koike K, et al. Late-onset cases
of familial hemophagocytic lymphohistiocytosis with
missense perforin gene mutations. Am J Hematol
2007;82:427-32. CrossRef
8. Hui WF, Luk CW, Chan WK, Miu TY, Yuen HL. Severe
lactic acidosis in an infant with haemophagocytic
lymphohistiocytosis. Hong Kong J Paediatr (New Series)
2012;17:183-9.
9. Chan JS, Shing MM, Lee V, Li CK, Yuen P. Haemophagocytic
lymphohistiocytosis in Hong Kong children. Hong Kong
Med J 2008;14:308-13.
10. Ho MH, Cheuk DK, Lee TL, Ha SY, Lau YL.
Haemophagocytic lymphohistiocytosis in Hong Kong
children have a wider clinical spectrum. Hong Kong Med J
2008;14:503-4.
11. Lee WI, Chen SH, Hung IJ, et al. Clinical aspects,
immunologic assessment, and genetic analysis in Taiwanese
children with hemophagocytic lymphohistiocytosis.
Pediatr Infect Dis J 2009;28:30-4. CrossRef
12. Chen RL, Hsu YH, Ueda I, et al. Cytophagic
histiocytic panniculitis with fatal haemophagocytic
lymphohistiocytosis in a paediatric patient with perforin
gene mutation. J Clin Pathol 2007;60:1168-9. CrossRef