Hong Kong Med J 2014;20:165–7 | Number 2, April 2014
DOI: 10.12809/hkmj133863
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Atypical focal cortical dysplasia in a patient
with Cowden syndrome
KM Cheung, MB, ChB1; CW Lam,
MB, ChB, PhD4; YK Chan, MB, BS2; WK Siu, MB,
BS5; L Yong, MB, ChB3
1 Department of
Paediatrics, Caritas Medical Centre, Shamshuipo, Hong Kong
2 Department of Medicine and Geriatrics,
Caritas Medical Centre, Shamshuipo, Hong Kong
3 Department of Surgery, Caritas Medical Centre, Shamshuipo, Hong Kong
4 Department of Pathology,
The University of Hong Kong, Pokfulam, Hong Kong
5 Kowloon West Cluster
Laboratory Genetic Service, Chemical Pathology Laboratory,
Department of Pathology, Princess Margaret Hospital, Laichikok,
Hong Kong
Corresponding author: Dr KM Cheung (jennykmcheung@gmail.com)

Abstract
A macrocephalic girl presented with generalised
epilepsy due to focal cortical dysplasia. She later
developed multiple hamartomatous lesions and was
diagnosed to have Cowden syndrome. The diagnosis
was confirmed by identification of a novel frameshift
mutation in the PTEN gene of the patient.
Case report
A 10-year-old Chinese girl presented to a
paediatric clinic with epilepsy. She had normal intelligence and
social interaction ability, and no relevant family history. She
had been having recurrent sleep seizures, with generalised
twitching of all four limbs, cyanosis, up-rolling of eyeballs,
drooling, and urinary incontinence. Her head circumference was 61
cm (5 cm > the 97th percentile). Physical examination was
otherwise normal. The electroencephalogram showed an abnormal
focus at the right temporo-occipital region. Goldmann perimetry
revealed no abnormality. Magnetic resonance imaging (MRI) with
contrast showed pachygyria (total loss of sulcation) with
hyperplastic white matter and disorganisation of the grey and
white matters in the right occipital lobe. A 2.5 cm x 1.8 cm T2
hyperintense area around the right occipital horn was consistent
with gliosis. The right occipital lobe was mildly enlarged.
Repeated MRI 5 and 7 years after presentation showed no interval
changes. Her findings were compatible with focal cortical
dysplasia involving the right occipital lobe (Fig
1). The epilepsy was under good control with carbamazepine
treatment. She developed a goitre at the age of 16 years, but was
euthyroid and ultrasonography showed a multinodular goitre.
Hypertrichosis was noted when she was 17 years old. Multiple tiny
papules were noted at the perinasal region, of which the patient
regarded as coarse skin. At the age of 19 years, she was
incidentally found to have iron deficiency anaemia (haemoglobin 96
g/L [reference range, 117-149 g/L], mean corpuscular volume 68.5
fL [82-97 fL], serum iron 2 μmol/L [5.0-30.4 μmol/L], total iron
binding capacity 77.1 μmol/L [44.8-76.0 μmol/L]). She had no
gastro-intestinal symptoms, but did receive iron supplements.
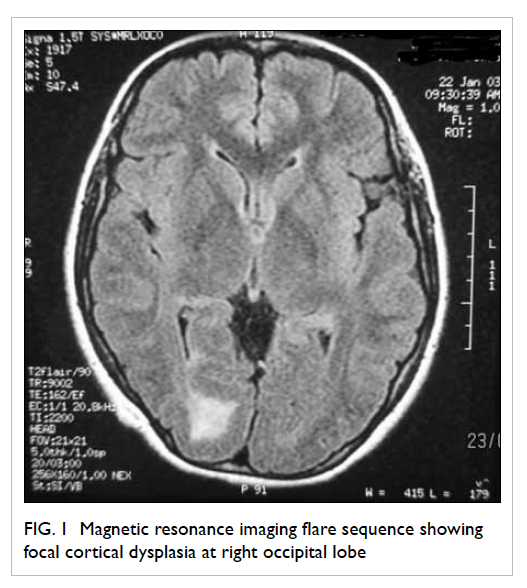
Figure 1. Magnetic resonance imaging flare sequence showing focal cortical dysplasia at right occipital lobe
She had a left hemithyroidectomy at the age
of 22 years for increasing size of a dominant left-sided thyroid
nodule that expanded from 1.7 × 1.4 × 0.8 cm to 3.6 × 2.9 × 4.2 cm
over 10 months. Fine-needle aspiration showed lymphocytic
thyroiditis, and excisional biopsy revealed adenomatous
hyperplasia. At the age of 23 years, enlargement of a thyroid
nodule in the right thyroid lobe (3 cm in diameter) was noted;
fine-needle aspiration suggested it was an adenomatous nodule.
Total thyroidectomy was performed, and excisional biopsy revealed
an atypical nodule with atypical enlarged vesicular nuclei and
small distinct nucleoli. The patient also developed a 3-cm scalp
papilloma, shown by excisional biopsy to be fibrous papule. At the
age of 24 years, she also experienced coffee ground vomiting;
oesophagogastroduodenoscopy showed diffuse glycogen deposits at
the lower oesophagus and multiple gastric polyps. Polypectomy
yielded lymphoid hyperplasia, and oesophageal mucosa biopsy was
reported as showing glycogenic acanthosis.
In view of her multiple benign tumours, the
possibility of a hamartomatous polyposis syndrome was suspected.
Subsequent clinical examination yielded multiple papillomas over
the face and tongue, but there was no pigmentation of the lips.
These mucocutaneous features were pathognomonic criteria of Cowden
syndrome. Notably, macrocephaly, thyroid adenoma,
gastro-intestinal hamartomas (oesophageal glycogenic acanthosis
and gastric polyps), and skin fibromas fulfilled one major and
three minor clinical criteria of Cowden syndrome. Mutational
analysis of the PTEN gene (Fig 2) showed a heterozygous thymine
deletion in exon 8 at position 1023 (NM_000314.4(PTEN_
v001):c.1023del). This deletion creates a frame shift starting at
codon Phe341. The new reading frame ends in a stop codon 2
position downstream (NM_000314.4(PTEN_i001):p.(Phe341Leufs*3).
This mutation was not detected in the mother. The patient’s father
was deceased 10 years earlier due to lung cancer, but he did not
have macrocephaly or a history of any other tumours.
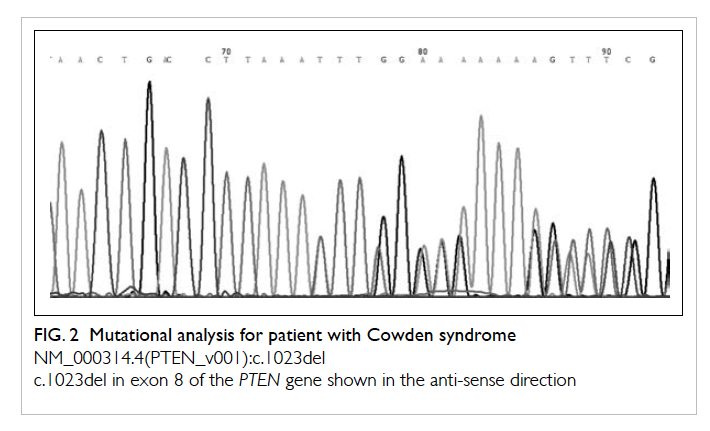
Figure 2. Mutational analysis for patient with Cowden syndrome
Discussion
Cowden syndrome was first described by
Lloyd and Dennis in 1963.1
It is characterised by macrocephaly, mucocutaneous lesions, acral
(extremity/limb) keratosis, papillomatous papules, and high risk
of development of cancer in the breast, thyroid, and endometrium.
It is a rare autosomal dominant disease, with an estimated
prevalence of 1 in 1 000 000 to 250 000 based on genetic
identification.2 Lesions
can occur in tissues derived from all three embryonic germ cell
layers. The subtle and variable clinical manifestations contribute
to the difficulty in making a clinical diagnosis. In 1996, the
international Cowden Consortium identified germline PTEN
(phosphatase and tensin homologue on chromosome 10) mutations as a
cause of Cowden syndrome.2
This is a tumour suppressor gene encoding a major lipid
phosphatase that functions in the phosphoinositide 3-kinase
signalling cascade.3 It
regulates cellular processes crucial for normal development,
including cell proliferation, soma growth, cell death, and cell
migration.4 The importance
on PTEN mutations in making a diagnosis, genetic
counselling, and clinical surveillance for the development of
malignancies is well recognised. Apart from cancer susceptibility,
the PTEN mutation was also implicated as candidate gene in
developmental disorder like autism and mental retardation.4
Our patient was first suspected to have
Cowden syndrome because of the interesting endoscopic findings, as
multiple gastric polyps are rarely encountered in young adults.
Patients with such polyps should be examined for various genetic
syndromes associated with gastro-intestinal polyps, including
Peutz-Jeghers syndrome, Cowden syndrome, and Cronkhite-Canada
syndrome.5 Histology shows
Helicobacter pylori–negative, non-specific lymphoid
hyperplasia. Diffuse oesophageal glycogenic acanthosis has seldom
been seen in young adults. The concurrence of oesophageal
glycogenic acanthosis and multiple gastric polyps is associated
with Cowden syndrome.6 7 8
Adult-onset Lhermitte-Duclos disease (LDD),
which presents clinically with progressive cerebellar sign and
increased intracranial pressure, is also a feature. A dysplastic
cerebellar gangliocytoma is now considered to be one of the
central nervous system (CNS) pathognomonic criteria by the revised
Cowden Syndrome Consortium. The other overt CNS manifestations of
Cowden syndrome included macrocephaly, autism, developmental
delay, brain tumour, and LDD. The unique feature in our patient
was the presentation with generalised epilepsy in childhood and
the identification of focal cortical dysplasia in the right
occipital region. To our knowledge, such brain MRI imaging has not
been described previously in Cowden syndrome.9 Apart from regulating cell growth, the PTEN
gene also plays a role in cell migration. It interacts with focal
adhesion kinase, which results in the inhibition of cell migration
and spread. Focal cortical dysplasia is a kind of neuronal
migration disorder. Our patient was noted to have a right
occipital focal cortical dysplasia and right megalencephaly. These
features suggest CNS manifestations of the PTEN mutation.
Inclusion of this feature in Cowden syndrome may facilitate early
diagnosis.
Cowden syndrome represents a late-onset
phenotype of the PTEN mutation. Early presentation of PTEN mutations
include Bannayan-Riley-Ruvalcaba syndrome and autism spectrum
disorder with macrocephaly. Bannayan-Riley-Ruvalcaba syndrome is a
congenital disorder characterised by macrocephaly, hamartomatous
intestinal polyps, lipomas, and pigmented macules on the penis. To
our knowledge, our patient represents a new PTEN mutation
phenotype, with macrocephaly and focal cortical dysplasia at the
occipital region and subsequent full-blown presentation of Cowden
syndrome.
In conclusion, we believe we have
identified a new phenotype of Cowden syndrome and a new PTEN
indel mutation. This PTEN mutation appears to cause
megalencephaly, epilepsy, focal cortical dysplasia in the
occipital region in childhood, and multiple hamartomatous lesions
in late adolescence and early adulthood. The inclusion of focal
cortical dysplasia in the occipital region as a CNS feature of the
PTEN mutation could facilitate early diagnosis of PTEN
mutation syndromes, and clinicians to undertake early surveillance
for possible malignancies. In the latest published prospective
study of the Ohio cohort of patients with germline PTEN
mutation, the penetrance of breast cancer was found to begin at
around the age of 30 years rising to an estimated 85% lifetime
risk.10 The lifetime risk
of breast cancer in females with PTEN mutations was even
higher than the best estimates for individuals with BRCA1
or BRCA2 mutations.11
The PTEN-related endometrial cancer risk begins at the age
of 25 years rising to 30% by the age of 60 years.10 The thyroid cancer risk begins at birth and
continues lifelong.10
Risks of colorectal and kidney cancers begin at around the age of
40 years, with a lifetime risk of 9% and 34%, respectively.10 The earliest reported age of onset of
melanoma was in a 3-year-old patient.10
References
1. Lloyd KM 2nd, Dennis M. Cowden's
disease. A possible new symptom complex with multiple system
involvement. Ann Intern Med 1963;58:136-42. CrossRef
2. Farooq A, Walker LJ, Bowling J,
Audisio RA. Cowden syndrome. Cancer Treat Rev 2010;36:577-83. CrossRef
3. Shen WH, Balajee AS, Wang J, et
al. Essential role for nuclear PTEN in maintaining chromosomal
integrity. Cell 2007;128:157-70. CrossRef
4. Orrico A, Galli L, Buoni S, Orsi
A, Vonella G, Sorrentino V. Novel PTEN mutations in
neurodevelopmental disorders and macrocephaly. Clin Genet
2009;75:195-8. CrossRef
5. Hizawa K, Iida M, Matsumoto T,
et al. Gastrointestinal manifestations of Cowden's disease. Report
of four cases. J Clin Gastroenterol 1994;18:13-8. CrossRef
6. McGarrity TJ, Wagner Baker MJ,
Ruggiero FM, et al. GI polyposis and glycogenic acanthosis of the
esophagus associated with PTEN mutation positive Cowden syndrome
in the absence of cutaneous manifestations. Am J Gastroenterol
2003;98:1429-34. CrossRef
7. Vasovcak P, Krepelova A,
Puchmajerova A, et al. A novel mutation of PTEN gene in a patient
with Cowden syndrome with excessive papillomatosis of the lips,
discrete cutaneous lesions, and gastrointestinal polyposis. Eur J
Gastroenterol Hepatol 2007;19:513-7. CrossRef
8. Ha M, Chung JW, Hahm KB, et al.
A case of Cowden syndrome diagnosed from multiple gastric
polyposis. World J Gastroenterol 2012;18:861-4. CrossRef
9. Lok, C, Viseux V, Avril MF, et
al. Brain magnetic resonance imaging in patients with Cowden
syndrome. Medicine (Baltimore) 2005;84:129-36. CrossRef
10. Tan MH, Mester JL, Ngeow J,
Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals
with germline PTEN mutations. Clin Cancer Res 2012;18:400-7. CrossRef
11. Metcalfe K, Lubinski J, Lynch
HT, et al. Family history of cancer and cancer risks in women with
BRCA1 or BRCA2 mutations. J Natl Cancer Inst 2010;102:1874-8. CrossRef