Hong Kong Med J 2014;20:63–6 | Number 1, February 2014
DOI: 10.12809/hkmj133826
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome: a treatable genetic liver disease warranting urgent diagnosis
Hencher HC Lee, MA, FRCPA1;
KH Poon, FHKAM (Paediatrics)2;
CK Lai, MSc1;
KM Au, MSc1;
TS Siu, MPhil3;
Judy PS Lai, MSc4;
Chloe M Mak, PhD, FHKAM (Pathology)1;
YP Yuen, MSc, FHKAM (Pathology)1;
CW Lam, PhD, FHKAM (Pathology)5;
Albert YW Chan, MD, FHKAM (Pathology)1
1 Department of Pathology, Princess Margaret Hospital, Laichikok, Hong
Kong
2 Department of Paediatrics and Adolescent Medicine, Tuen Mun Hospital,
Tuen Mun, Hong Kong
3 Division of Clinical Biochemistry, Queen Mary Hospital, Pokfulam, Hong
Kong
4 Department of Clinical Pathology, Tuen Mun Hospital, Tuen Mun, Hong
Kong
5 Department of Pathology, The University of Hong Kong, Queen Mary
Hospital, Pokfulam, Hong Kong
Corresponding author: Dr CW Lam (ching-wanlam@pathology.hku.hk)

Abstract
Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is an autosomal
recessive disorder caused by a defect in ornithine
translocase. This condition leads to variable
clinical presentations, including episodic
hyperammonaemia, hepatic derangement, and
chronic neurological manifestations. Fewer than 100
affected patients have been reported worldwide. Here
we report the first two cases in Hong Kong Chinese,
who were compound heterozygous siblings for
c.535C>T (p.Arg179*) and c.815C>T (p.Thr272Ile)
in the SLC25A15 gene. When the mother refused
prenatal diagnosis for the second pregnancy, urgent
genetic testing provided the definitive diagnosis
within 24 hours to enable specific treatment.
Optimal management of these two patients relied on
the concerted efforts of a multidisciplinary team and
illustrates the importance of an expanded newborn
screening service for early detection and treatment
of inherited metabolic diseases.
Introduction
The urea cycle is the major pathway of nitrogen
metabolism in the human body. Excess nitrogen, in
the form of ammonia, is converted via this cycle to
urea and excreted through the kidneys. In humans,
the cycle entails five key enzymes, including
carbamoyl-phosphate synthetase I (CPS1), ornithine
transcarbamylase (OTC), argininosuccinate
synthetase, argininosuccinate lyase, and arginase;
while an additional enzyme named N-acetylglutamate
synthase provides CPS1 with its essential cofactor.1
A defect in any of these six enzymatic pathways or
the two associated transporters, namely citrin and
ornithine translocase, causes urea cycle disorders.2
Patients with complete deficiency of the affected
enzyme present with significant hyperammonaemia
in the neonatal period. It is a serious and often
lethal condition or causes irreversible brain damage
especially when the diagnosis or treatment is delayed
or ineffective. On the other hand, patients with partial
enzyme deficiencies or defective transporters can
present later in life, from infancy to adulthood, and
manifest whenever the urea cycle is overwhelmed by environmental triggers or stresses. These result in
acute hyperammonaemic episodes.2
Hyperornithinaemia-hyperammonaemia-homocitrullinuria
syndrome (HHH syndrome;
MIM#238970) is an autosomal recessive disorder
caused by a defect in ornithine translocase
(SLC25A15 or ORNT1, MIM*603861). The disorder
is exceedingly rare; with fewer than 100 patients
having been reported worldwide, although its
incidence in northern Saskatchewan in Canada
was estimated to be 1 in 1500 (with a carrier rate
of 1 in 19).3 The syndrome was first described by
Shih et al4 in 1969 with a neurological phenotype
entailing seizures and mental retardation. It was
later found that the clinical presentations of HHH
syndrome can be highly variable, and include spastic
paraplegia, pyramidal and extrapyramidal signs,
stroke-like episodes, hypotonia, seizures, ataxia,
protein intolerance, failure to thrive, and hepatic
failure.5 6 7 Liver biopsies typically reveal vacuolated
hepatocytes distended with glycogen on light
microscopy and bizarre-looking mitochondria on
electronic microscopy.8 So far, no definite genotype-phenotype
correlation has been noted, with a high degree of clinical heterogeneity even among patients
harbouring the same genetic defect.9 However,
patients with HHH syndrome can respond well to a
low-protein diet with improvements in neurological
symptoms and hepatic function, for which reason
an accurate diagnosis is critical to management.5 10
For the first time in Hong Kong, here we describe
HHH syndrome in a pair of siblings and their clinical,
biochemical, and molecular profiles, with a view
to facilitate understanding of this disorder in our locality. The diagnosis of this family also revealed a
possible founder mutation in ethnic Chinese.
Case report
The patient was an ethnic Han Chinese boy, born
healthy to a non-consanguineous couple and fed on
both human and formula milk in 2007. He presented
with neonatal jaundice with serum bilirubin up to 338 µmol/L (diazo method) or 295 µmol/L
(photometric method) on day 5, which dropped to 179 µmol/L (photometric method) on day 6 after phototherapy.
He was then discharged without further blood taking,
since otherwise he was clinically well. However, 1
month later he presented with persistent jaundice
and a liver palpable 2 cm below the costal margin but
no clinical splenomegaly. The total bilirubin was 99 µmol/L with a direct bilirubin of 27 (reference range
[RR], 1-5) µmol/L and an alkaline phosphatase (ALP) of 529 (RR, 82-383) U/L. His γ-glutamyltransferase (GGT) ranged from 345 to 388 (reference level, <220) U/L but the alanine transaminase (ALT) was normal
at 32 to 35 (RR, 4-35) U/L. While his bilirubin and
GGT levels gradually normalised at 2 months, even
at 12 months the ALT remained elevated at 311 U/L
and the ALP was 418 U/L (RR, 104-345 U/L). A
deranged clotting profile with a prothrombin time of
20.5 (RR, 10.4-12.6; international normalised ratio,
2.0) seconds, an activated partial thromboplastin time
of 36.3 (RR, 26.4-35.3) seconds, and a serum bile acid
level of 10.8 (reference level, <7) μmol/L were noted.
At this juncture, his liver remained palpable, 1 cm
below the costal margin. In addition, he was noted
to have alpha thalassaemia trait.
At the age of 11 months, gas chromatography–mass spectrometry of urine detected significant
hyperexcretion of uracil and moderately excessive
excretion of orotic acid, while he was taking an
unrestricted protein diet (approximately 3 g/kg/day).
He also had homocitrullinuria of up to 71 (reference
level, <9) μmol/mmol creatinine, which was also
demonstrated by liquid chromatography–tandem
mass spectrometry. Plasma amino acids analysis by
high-performance liquid chromatography detected
excessive alanine, arginine, ornithine, and
methionine concentrations, postprandially (Fig).
At the age of 12 months, the blood ammonia was elevated at 132 (RR, 16-60) µmol/L, the simultaneous
blood glucose was 3.8 mmol/L, and the lactate was 2.5
(RR, 0.5-2.2) mmol/L. The biochemical picture was
suggestive of urea cycle dysfunction. Interestingly,
at that juncture he was clinically well and had no
vomiting or encephalopathy. His blood ammonia
decreased to 59 µmol/L on rechecking after 24
hours just before institution of protein restriction
(0.9 g/kg/day); over the next 14 days it fluctuated
between 43 and 84 µmol/.
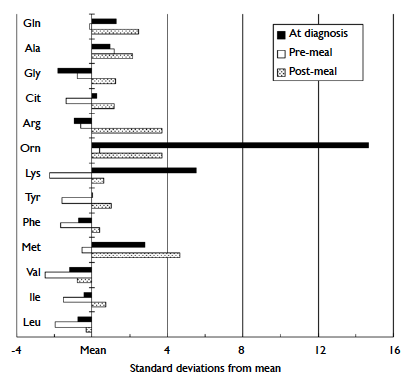
Figure. The amino acid profile of the proband on different occasions
Mutational analysis was performed by
polymerase chain reaction and Sanger sequencing with genomic DNA. While no mutation was noted
in the OTC gene, the patient was shown to be
heterozygous for two different mutations, c.535C>T
(p.Arg179*) and c.815C>T (p.Thr272Ile), in the SLC25A15 gene. The latter missense mutation was
not found in 100 Chinese control chromosomes
tested. Compound heterozygosity was confirmed by
analysing the parental DNA.
The patient's liver became impalpable 1 month
after therapy. Normalisation of the serum ALT level
was noted 1 month after treatment, although plasma
ornithine and urine orotic acid levels remained
elevated. Coagulation factor VII and X levels were
normal during convalescence. At the age of 6 years,
the boy had no acute encephalopathy or pyramidal
signs, but did exhibit mild clumsiness and subtle gait
ataxia (only evident on tandem walking).
The mother became pregnant 1 year later in
2009, when the proband was 2 years old. In view of
the family history of HHH syndrome, counselling was
provided by the obstetrician early during gestation, yet
the parents opted not to obtain a prenatal diagnosis.
Prior arrangement was then made with the chemical
pathologist to have a semi-urgent molecular diagnosis
to facilitate therapy for the neonate if necessary.
This younger brother was immediately started on a
low protein (≤1.2 g/kg/day) diet, which consisted of
breastfeeding and a zero-protein formula after delivery.
Aged 12 hours, the postprandial blood ammonia was
68 µmol/L (reference level, <100 µmol/L) and blood
for molecular genetics was sampled simultaneously.
The boy only had physiological jaundice and no
other signs, but was also soon confirmed to have a
compound heterozygous form of the two familial
mutations about which the paediatrician was notified
within 24 hours of blood sampling. In view of the
prompt definitive diagnosis, protein restriction was
continued with confidence. His ammonia peaked
at 111 µmol/L and then normalised, whilst his
ALT level remained normal in the neonatal period.
His coagulation factors VII and X levels were also
normal. The boy did not have any episodes of acute
encephalopathy and developed normally when seen
for follow-up at the age of 3 years.
Discussion
When arginase cleaves arginine in the last step of
the urea cycle to produce urea and ornithine, the
ornithine translocase enzyme transports cytosolic
ornithine back into the mitochondria for subsequent
urea cycles in exchange of mitochondrial citrulline.11
The gene SLC25A15 was cloned in 1999 and found
to account for the HHH syndrome,12 and molecular
modelling was reported early in 2012.13 A founder
mutation p.Phe188del was reported in French-Canadian patients,12 and in Japanese patients the
p.Arg179* mutation was also noted to be frequent.14 15
The nonsense mutation p.Arg179* variant is predicted to cause premature termination of the
protein. Although common in Japan,14 it was also
reported in other ethnic groups.15 The missense
mutation p.Thr272Ile was reported in 2009 by
Tessa et al15 in one Taiwanese patient, with recently
published functional proof of its pathogenicity.
However, this missense mutation has never been
reported in other ethnic groups. We therefore
postulate that it could be a common mutation,
possibly having an ancestral founder gene effect in
ethnic Chinese. If this is confirmed in more patients
of Chinese ethnicity, it may aid prioritising workflow
for the genetic testing of individuals suspected to
have HHH syndrome. In which case, they could
undergo more focused molecular investigation
instead of whole gene sequencing. Consequently, a
more rapid diagnosis could enable more prompt and
appropriate treatment.
To the best of our knowledge, these were
the first two cases of HHH syndrome reported in
Hong Kong. The proband’s metabolic profile in
early infancy was particularly illustrative of the
natural course of the associated hepatic disease.
The untreated first child had pronounced neonatal
jaundice which responded to phototherapy and soon
evolved into mild transient hyperbilirubinaemia
with an accompanying elevation in serum ALP
but not ALT levels in early infancy. Subsequently,
despite resolution of jaundice, he showed moderate
hepatocellular derangement and dysfunction with
a coagulopathy and hyperammonaemia, which
responded to protein restriction. The younger
brother had no serum ALT level elevation while the
ammonia level was only mildly raised in the first week
of life, at which time he was proactively commenced
on protein restriction. These two cases demonstrate
that metabolic profiling, including the ammonia
level, should be included in the initial workup for any
infant with unexplained prolonged liver dysfunction
and may provide a clue to a possible underlying
defect in the urea cycle. The HHH syndrome is rare,
yet a readily treatable cause to consider in Chinese
patients with unusual plasma amino acid patterns. In
addition, modern medical technologies (eg tandem
mass spectrometry) allow multiplex screening of
classical inherited metabolic disorders that can
detect HHH syndrome using hyperornithinaemia as
the disease marker.16 The successful diagnosis and
management of these siblings entailed a concerted
effort and collaboration of a multidisciplinary team.
Notably, the diagnosis of rare diseases is often
difficult, and the importance of having an integrated
pathology service is crucial.
Prenatal diagnosis for the younger brother was
possible but declined by the parents, making timely
intervention of the chemical pathology laboratory
even more critical for establishing or excluding the
diagnosis in the neonate. In this clinical setting, a rapid and definitive diagnosis (within 24 hours)
provided by genetic testing was important as a
mildly elevated ammonia level in an asymptomatic
newborn may be hard to interpret. It allowed the
clinicians to counsel the parents accordingly on the
need for lifelong protein restriction to minimise
the chance of decompensation. Although late-onset
long-term neurological sequelae may not be
preventable,9 it is prudent to keep the two children
metabolically stable as far as possible, to mitigate
brain damage from decompensation.
In conclusion, HHH syndrome, although very
rare, is an inborn error of metabolism that can occur
in the Chinese and is readily detectable by tandem
mass spectrometry.17 If this technique could be
introduced to support a local newborn screening
programme, many more possibly treatable metabolic
disorders may be picked up.
References
1. Häberle J. Clinical practice: the management of hyperammonemia. Eur J Pediatr 2011;170:21-34. Crossref
2. Mitchell S, Ellingson C, Coyne T, et al. Genetic variation in the urea cycle: a model resource for investigating key candidate genes for common diseases. Hum Mutat 2009;30:56-60. Crossref
3. Sokoro AA, Lepage J, Antonishyn N, et al. Diagnosis and high incidence of hyperornithinemia-hyperammonemia-homocitrullinemia (HHH) syndrome in northern Saskatchewan. J Inherit Metab Dis 2010;33 Suppl 3:275-81. Crossref
4. Shih VE, Efron ML, Moser HW. Hyperornithinemia, hyperammonemia, and homocitrullinuria. A new disorder of amino acid metabolism associated with myoclonic seizures and mental retardation. Am J Dis Child 1969;117:83-92. Crossref
5. Al-Hassnan ZN, Rashed MS, Al-Dirbashi OY, Patay Z, Rahbeeni Z, Abu-Amero KK. Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome with stroke-like imaging presentation: clinical, biochemical and molecular analysis. J Neurol Sci 2008;264:187-94. Crossref
6. Lemay JF, Lambert MA, Mitchell GA, et al. Hyperammonemia-hyperornithinemia-homocitrullinuria syndrome: neurologic, ophthalmologic, and neuropsychologic examination of six patients. J Pediatr 1992;121:725-30. Crossref
7. Gatfield PD, Taller E, Wolfe DM, Haust MD. Hyperornithinemia, hyperammonemia, and homocitrullinuria associated with decreased carbamyl phosphate synthetase I activity. Pediatr Res 1975;9:488-97. Crossref
8. Haust MD, Gordon BA. Ultrastructural changes in the mitochondria in disorders in ornithine metabolism. Pediatr Res 1980;14:1411. Crossref
9. Debray FG, Lambert M, Lemieux B, et al. Phenotypic variability among patients with hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome homozygous for the delF188 mutation in SLC25A15. J Med Genet 2008;45:759-64. Crossref
10. Gjessing LR, Lunde HA, Undrum T, Broch H, Alme A, Lie SO. A new patient with hyperornithinaemia, hyperammonaemia and homocitrullinuria treated early with low protein diet. J Inherit Metab Dis 1986;9:186-92. Crossref
11. Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 2004;447:689-709. Crossref
12. Camacho JA, Obie C, Biery B, et al. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet 1999;22:151-8. Crossref
13. Wang JF, Chou KC. Insights into the mutation-induced HHH syndrome from modeling human mitochondrial ornithine transporter-1. PLoS One 2012;7:e31048. Crossref
14. Miyamoto T, Kanazawa N, Kato S, et al. Diagnosis of Japanese patients with HHH syndrome by molecular genetic analysis: a common mutation, R179X. J Hum Genet 2001;46:260-2. Crossref
15. Tessa A, Fiermonte G, Dionisi-Vici C, et al. Identification of novel mutations in the SLC25A15 gene in hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome: a clinical, molecular, and functional study. Hum Mutat 2009;30:741-8. Crossref
16. Chace DH, Kalas TA, Naylor EW. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin Chem 2003;49:1797-817. Crossref
17. Lee HC, Mak CM, Lam CW, et al. Analysis of inborn errors of metabolism: disease spectrum for expanded newborn screening in Hong Kong. Chin Med J (Engl) 2011;124:983-9.