Hong Kong Med J 2025;31:Epub 5 Dec 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Van der Woude syndrome with novel variants: a case series
LT Leung, MB, ChB; Stephanie KL Ho, MB, BS; WC Yiu, MSc; Min Ou, MPhil; Jennifer YY Poon, MB, ChB; Shirley SW Cheng, MB, ChB; Ivan FM Lo, MB, ChB; HM Luk, MD (HK)
Department of Clinical Genetics, Hong Kong Children’s Hospital, Hong Kong SAR, China
# Equal contribution
Corresponding author: Dr HM Luk (lukhm@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Case presentations
In January 2024, individuals presenting to the
Department of Clinical Genetics at Hong Kong
Children’s Hospital with suspected pathogenic
variants in the IRF6 or GRHL3 genes were assessed.
Eight patients with molecularly confirmed Van
der Woude syndrome (VWS) from four unrelated
families were identified, aged between 17 and 58
years, with a male-to-female ratio of 5:3. All four
index cases had an affected parent. Pedigrees of the
four families are shown in online supplementary Figure 1.
Cleft palate was observed in 75% (n=6) of
individuals, of whom two had bilateral cleft palate
and two had submucous cleft palate. Lower lip pits
were present in 62.5% (n=5). Cleft lip and/or alveolus
was evident in four patients (50%), usually affecting
both sides. Bifid uvula was observed in only one
individual, while hypodontia was seen in two. One
patient had ankyloglossia and two patients (25%)
developed vitiligo.
The clinical findings are summarised in the
Table. Clinical photos demonstrating oral findings
from Family 4 are shown in the Figure.
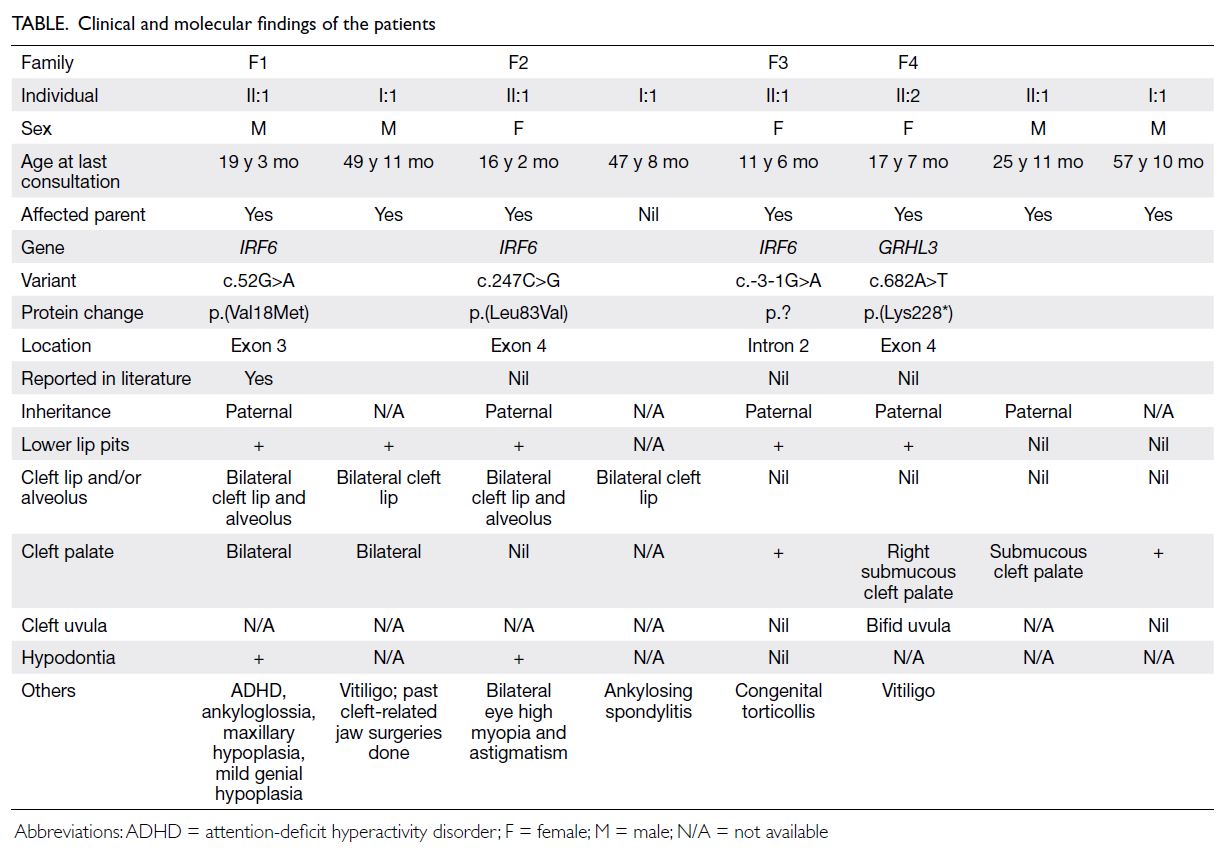
Table. Clinical and molecular findings of the patients
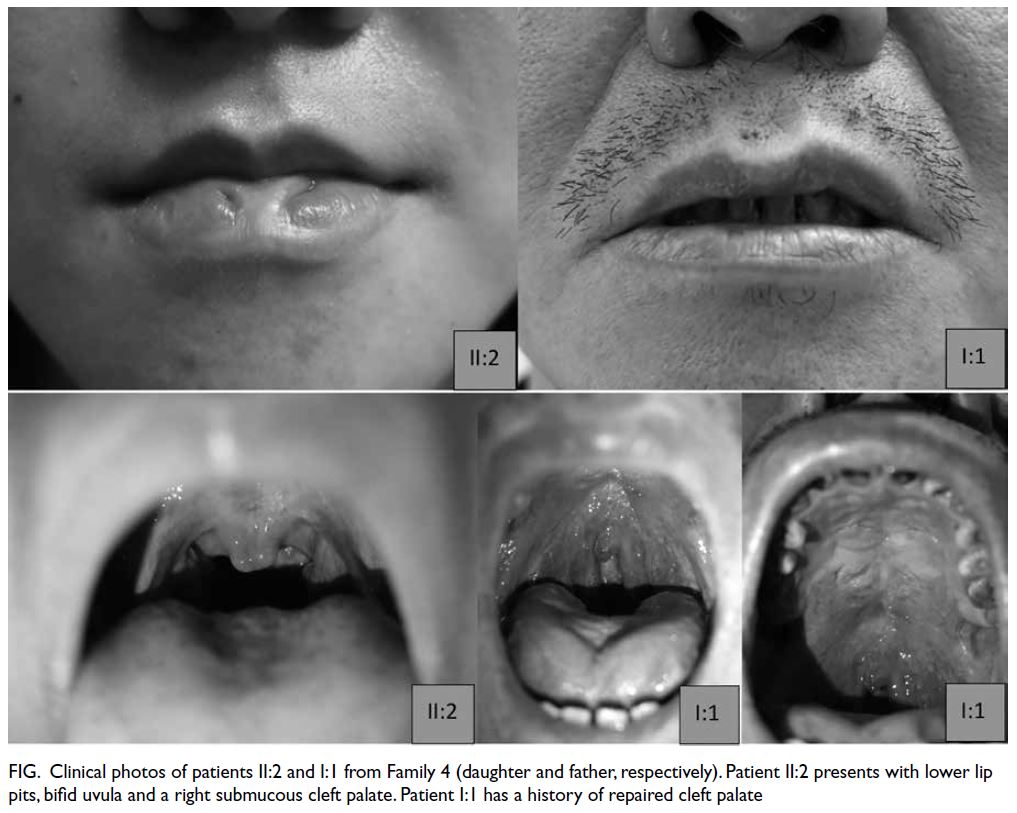
Figure. Clinical photos of patients II:2 and I:1 from Family 4 (daughter and father, respectively). Patient II:2 presents with lower lip pits, bifid uvula and a right submucous cleft palate. Patient I:1 has a history of repaired cleft palate
A different variant was identified in each of the
index patients in our cohort. Three harboured an IRF6
(NM_006147.4) variant, while the remaining index
patient had a variant in GRHL3 (NM_198173.3).
All variants were classified as likely pathogenic or
pathogenic according to the American College of
Medical Genetics and Genomics guidelines1. Two
of the IRF6 variants were missense variants, while
the remaining one was a splice site variant. The
only GRHL3 variant identified in our cohort was a
nonsense variant. All other variants in our cohort
were novel, except IRF6 c.52G>A p.(Val18Met)
which has been previously reported.2
The molecular findings of our patients are
summarised in the Table and online supplementary Figure 2.
Discussion
Orofacial cleft is a prevalent congenital defect with an estimated global occurrence of 1 in 500 to 1000 births and a comparatively higher incidence in
Asia according to 2019 Global Burden of Disease
data3. It is linked to both environmental and
genetic factors. Most cases are non-syndromic,
presenting a reduced risk of a genetic disorder, with
orofacial clefts manifesting as isolated structural
anomalies. Nevertheless, syndromic cases comprise
approximately 30% of all cases, with an associated
higher risk in patients with central cleft lip and/or
palate (CL/P) or isolated cleft palate.4 Cases may be
familial or non-familial. According to a local study,4
the diagnostic yield of genomic variants, including
variants of uncertain significance, for non-syndromic
orofacial clefts was only about 4%. In contrast, for
syndromic cases, the detection rate of genomic
variants is as high as around 70%. Among these,
most variants have been detected by karyotyping or
chromosomal microarray.5 In view of this finding,
genetic testing (ie, chromosomal microarray) is
conventionally offered as first-line screening in
patients with syndromic orofacial clefts. Whole
exome sequencing is less commonly performed in
Hong Kong’s public clinical sector, although there is
no international consensus. Clinical and molecular
findings from our four families with VWS suggest
that whole exome sequencing may be helpful in
making a genetic diagnosis in patients with orofacial
clefts. This is advantageous for both the patient
and their family, as reproductive options such
as preimplantation genetic diagnosis or prenatal
diagnosis can be provided for at-risk individuals.
Van der Woude syndrome has historically been
linked to pathogenic variants in IRF6, which encodes a
transcription factor essential for the differentiation of
skin, as well as breast and oral epithelium. Abnormal
differentiation of the epidermis or oral periderm
may be implicated in the pathogenesis of CL/P.
The subsequent increase in the number of patients
with CL/P has been accompanied by the discovery
of a second gene, GRHL3. In vivo studies have
demonstrated that GRHL3 regulates the epidermal
permeability barrier through action downstream
of IRF6, explaining the phenotypic convergence.6
Although it has been postulated that GRHL3 is
more likely associated with cleft palate and less likely with cleft lip, CL/P and lip pits, the number of
affected patients remains too small to draw definitive
conclusions about genotype-phenotype correlations.
In our cohort, all three individuals with a GRHL3
variant had cleft palate (mostly submucous), no cleft
lip, and only one had lower lip pits. These findings
appear to align with previous reports.2 4 5
According to the literature, approximately 60%
of VWS cases show familial occurrence.7 All index
individuals in our cohort had an affected parent.
The concurrence of lower lip pits and CL/P has
been reported as the most common clinical features
among patients with VWS, affecting 80% of cases.8
In total, 75% and 62.5% of our patients had cleft
palate or lower lip pits, respectively. Cleft lip and/or alveolus was evident in 50% of individuals. These
findings are comparable with figures described in
previous studies.2 4 5 Disease-causing variants in
both genes have also been associated with dental
anomalies, including hypodontia, dental aplasia,
and malocclusion. Bifid uvula, hypodontia and
ankyloglossia were also observed in our cohort. It
is known that individuals with VWS exhibit highly
variable expressivity, ranging from isolated lower lip pits to bilateral CL/P. As highlighted by Family
4 in our cohort, lower lip pits were identified only
in individual II:2, but not in her elder sibling (II:1)
or father (I:1). This again demonstrates the broad
intrafamilial variability.
An enriched prevalence of vitiligo was also
observed in our cohort. Although not previously
reported in patients with VWS, two patients (one
harbouring an IRF6 and one a GRHL3 variant) in
our cohort had vitiligo. In individual F1 I:1, vitiligo
was diagnosed during adulthood; in individual F4
II:2, at 16 years of age. Our observation points to
a possible disease association, with the argument
that interferon regulatory factors play a significant
role in the immune system by functioning as major
transcriptional regulators of type I interferon.9 Further
research has revealed that IRF6 regulates a subset of
toll-like receptor 3 responses in human keratinocytes
and may play a role in keratinocyte and/or immune
cell functions during cell damage and wound healing.
Alternatively, GRHL3 is a transcription factor critical
for epidermal differentiation and skin barrier repair.
A possible association of dysfunction in interferon
regulatory factors or GRHL3 with autoimmune dermatological conditions such as vitiligo cannot be
excluded. Further studies are required to confirm a
potential correlation.
All IRF6 and GRHL3 variants found in our
cohort, except one, were novel. In previous VWS
reports, missense variants in IRF6 were mainly
clustered in exons 3, 4, 7, and 9, whereas truncating
variants were evenly distributed across the whole
gene.10 One novel IRF6 variant in our cohort was
a missense variant in exon 4, while another was a
splice site variant in intron 2. No specific genotype-phenotype
correlation was established in our current
study due to the limited sample size.
Due to the highly variable expressivity and
incomplete penetrance, it is essential for clinicians
to remain vigilant in diagnosing individuals with
a relatively mild phenotype. Referral to clinical
geneticists for consideration of genetic testing is
beneficial for affected individuals with familial
occurrence of CL/P or when other features suggest a
syndromic diagnosis. In view of the limited number
of individuals identified in our cohort, future studies
may be needed to establish clearer genotype-phenotype genotypephenotype
correlations, explore a potential
association with vitiligo, and evaluate the diagnostic
efficacy of whole exome sequencing.
Author contributions
Concept or design: LT Leung, SKL Ho.
Acquisition of data: WC Yiu, M Ou, JYY Poon, SSW Cheng, IFM Lo, HM Luk.
Analysis or interpretation of data: LT Leung, SKL Ho, WC Yiu.
Drafting of the manuscript: LT Leung, SKL Ho, IFM Lo, HM Luk.
Critical revision of the manuscript for important intellectual content: IFM Lo, HM Luk.
Acquisition of data: WC Yiu, M Ou, JYY Poon, SSW Cheng, IFM Lo, HM Luk.
Analysis or interpretation of data: LT Leung, SKL Ho, WC Yiu.
Drafting of the manuscript: LT Leung, SKL Ho, IFM Lo, HM Luk.
Critical revision of the manuscript for important intellectual content: IFM Lo, HM Luk.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors disclosed no conflicts of interest.
Acknowledgement
The authors thank the patients and their family for their support.
Funding/support
This study received no specific grant from any funding agency
in the public, commercial, or not-for-profit sectors.
Ethics approval
This study was conducted in accordance with the Declaration
of Helsinki. The patients/their legal guardian provided written
informed consent for participation and publication of this
case report.
Supplementary material
The supplementary material was provided by the authors
and some information may not have been peer reviewed.
Accepted supplementary material will be published as
submitted by the authors, without any editing or formatting.
Any opinions or recommendations discussed are solely those
of the author(s) and are not endorsed by the Hong Kong
Academy of Medicine or the Hong Kong Medical Association.
The Hong Kong Academy of Medicine and the Hong Kong
Medical Association disclaim all liability and responsibility
arising from any reliance placed on the content.
References
1. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J,
Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K.
Standards and guidelines for the interpretation of
sequence variants: a joint consensus recommendation of
the American College of Medical Genetics and Genomics
and the Association for Molecular Pathology. Genetics in
medicine. 2015 May;17(5):405-23. Crossref
2. Kondo S, Schutte BC, Richardson RJ, et al. Mutations
in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet 2002;32:285-9. Crossref
3. Wang D, Zhang B, Zhang Q, Wu Y. Global, regional and
national burden of orofacial clefts from 1990 to 2019:
an analysis of the Global Burden of Disease Study 2019.
Annals of Medicine. 2023 Dec 12;55(1):2215540. Crossref
4. Chan KW, Lee KH, Pang KK, Mou JW, Tam YH. Clinical
characteristics of children with orofacial cleft in a
tertially centre in Hong Kong. HK J Paediatr (new series)
2013;18:147-51.
5. Li YY, Tse WT, Kong CW, et al. Prenatal diagnosis and
pregnancy outcomes of fetuses with orofacial cleft: a
retrospective cohort study in two centres in Hong Kong.
Cleft Palate Craniofac J 2024;61:391-9. Crossref
6. De La Garza G, Schleiffarth JR, Dunnwald M, Mankad A,
Weirather JL, Bonde G, Butcher S, Mansour TA, Kousa YA,
Fukazawa CF, Houston DW. Interferon regulatory factor
6 promotes differentiation of the periderm by activating
expression of Grainyhead-like 3. Journal of investigative
dermatology. 2013 Jan 1;133(1):68-77. Crossref
7. Children's Hospital of Philadelphia. Van der Woude
Syndrome [Internet]. Philadelphia: CHOP; 2024 Mar 31
[cited 2025 Nov 25]. Available from: https://www.chop.edu/conditions-diseases/van-der-woude-syndrome
8. Hersh JH, Verdi GD. Natal teeth in monozygotic twins
with Van der Woude syndrome. Cleft Palate Craniofac J
1992;29:279-81. Crossref
9. Honda K, Takaoka A, Taniguchi T. Type I interferon
[corrected] gene induction by the interferon regulatory
factor family of transcription factors. Immunity
2006;25:349-60. Crossref
10. Leslie EJ, Standley J, Compton J, Bale S, Schutte BC,
Murray JC. Comparative analysis of IRF6 variants in
families with Van der Woude syndrome and popliteal
pterygium syndrome using public whole-exome databases.
Genetics in Medicine. 2013 May;15(5):338-44. Crossref