Hong Kong Med J 2025;31:Epub 4 Aug 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Spectrum of inherited eye disorders at Hong Kong Children’s Hospital: insights into the local genetic landscape and experience with ocular genetic services
Shirley SW Cheng, MB, ChB, FHKAM (Paediatrics)1; Stephanie Cheung, FCOphthHK, FHKAM (Ophthalmology)2,3; TC Ko, FRCS (Edin), FCOphth3,4,5; TL Lee, MB, BS, FHKAM (Paediatrics)6; Jason Yam, FCOphthHK, FHKAM (Ophthalmology)2,3,7; HM Luk, MD, FHKAM (Paediatrics)1
1 Department of Clinical Genetics, Hong Kong Children’s Hospital, Hong Kong SAR, China
2 Clinical Services Department, Hong Kong Eye Hospital, Hong Kong SAR, China
3 Department of Ophthalmology, Hong Kong Children’s Hospital, Hong Kong SAR, China
4 Department of Ophthalmology, Tung Wah Eastern Hospital, Hong Kong SAR, China
5 Department of Ophthalmology, Pamela Youde Nethersole Eastern Hospital, Hong Kong SAR, China
6 Hospital Chief Executive Office, Hong Kong Children’s Hospital, Hong Kong SAR, China
7 Department of Ophthalmology and Visual Sciences, The Chinese University of Hong Kong, Hong Kong SAR, China
Corresponding author: Dr Jason Yam (yamcheuksing@cuhk.edu.hk); Dr HM Luk (lukhm@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Abstract
Introduction: Inherited eye disorders (IEDs) are a
leading cause of visual impairment. However, local
data and information about the genetic landscape
of IEDs in Hong Kong remain limited. This study
aimed to examine the diagnostic yield, mutational
spectrum, and clinical utility of genomic testing in
patients with IEDs at a major local centre.
Methods: This retrospective observational study
included 130 patients with suspected IEDs who
attended the genetic counselling clinic at the
Department of Clinical Genetics of the Hong
Kong Children’s Hospital between December 2021
and October 2023. Analyses were conducted on
the spectrum of ocular genetic disorders, genetic
variants, diagnostic yields, and clinical utility of
genomic testing.
Results: The overall diagnostic yield of genomic
testing was 51.5%. Inherited retinal disorders
accounted for approximately 60% of positive
results. Patients with syndromic features and a
positive family history were significantly more
likely to receive a molecular diagnosis (P<0.05).
Clinical utility of genomic testing was observed
in over 70% of patients with positive results.
With genetic counselling, a confirmed molecular
diagnosis contributed to disease prognostication,
avoided unnecessary investigations, guided clinical
management, and facilitated reproductive planning
and family cascade screening.
Conclusion: There is a growing demand for the application of genomic medicine in patients with
IEDs. Genetic testing is widely accepted and
demonstrates high diagnostic and clinical utilities.
The multidisciplinary team clinic service model
is the global trend for integrating genomic testing
into routine care. Hong Kong Children’s Hospital is
implementing this model to meet the evolving needs
of this patient population.
New knowledge added by this study
- The local diagnostic yield of genomic testing in patients with inherited eye disorder (IED) is 51.5%.
- Molecular confirmation of IEDs in more than 70% of patients demonstrated the clinical utility of genomic testing.
- Incorporation of genetic testing into routine IED workup is imperative.
- Implementation of a multidisciplinary team or combined clinic model—including ophthalmologists, geneticists, genetic counsellors, optometrists, and nurses—enables personalised and timely management of IED patients.
Introduction
According to World Health Organization estimates,
approximately 19 million children under the age
of 15 years are visually impaired, with 1.4 million exhibiting irreversible impairment.1 Among cases
of severe visual impairment diagnosed before the
age of 1 year, around one-third are attributable to
genetic causes.2
Substantial proportions of childhood and adult-onset
visual impairments are caused by inherited eye
disorders (IEDs), which include anterior segment
dysgenesis; inherited retinal disorders (IRDs);
microphthalmia, anophthalmia, and coloboma;
ocular tumours; congenital cataracts; and albinism.
Over the past three decades, more than 450 genes
have been associated with IEDs.2 3 Genetic diagnosis
in such cases is challenging due to both clinical and
genetic heterogeneity.
Ocular genetics has rapidly evolved over the
past decade—from identifying inheritance patterns
of IEDs to establishing genotype-phenotype
correlations for disease prognostication and enrolling
patients in gene therapy trials. In 2018, the United
States Food and Drug Administration approved the
first ocular gene therapy, Luxturna, for the treatment
of RPE65-related inherited retinal disease.4 In
2012, the American Academy of Ophthalmology
published diagnostic guidelines encouraging the
routine use of genetic testing for IEDs.5 Multiple
genes can now be assessed simultaneously through
a single genomic test, which is particularly useful for
identifying heterogeneous single-gene disorders and
resolving cases where a clinical diagnosis is difficult
to establish.6 Advances in sequencing technologies
are uncovering the molecular aetiologies of various
disorders. Consequently, the genomic approach to IEDs is gaining popularity, highlighting the
need for more sophisticated genomic testing and
comprehensive ocular genetic services.
In Hong Kong, the Retinitis Pigmentosa
Registry—the first of its kind among Chinese
populations globally—was established in 1995. Its
main objectives are to provide detailed ophthalmic
and genetic examinations for patients with inherited
retinal degenerative diseases and to build a database
for future scientific, medical, and sociological
research.7 However, local data remain limited and
the genetic landscapes of other IEDs are still unclear.
Hong Kong Children’s Hospital (HKCH)
serves as the tertiary referral centre for complex,
serious, and uncommon paediatric cases requiring
multidisciplinary management, providing diagnosis,
treatment, and rehabilitation services across the
territory. In 2021, the Clinical Genetics Service
Unit (CGSU) at HKCH was established as the
first clinical genetics branch under the Hospital
Authority. In July 2023, the Clinical Genetic Service
(CGS) of the Department of Health (DH)—the
former government-funded tertiary genetic referral
centre providing genetic counselling and laboratory
services to the entire Hong Kong population—was integrated with the CGSU and renamed the
Department of Clinical Genetics (DCG) under the
Hospital Authority. As a major clinical genetics
service provider in Hong Kong, the DCG now offers
genetic counselling services territory-wide.
Acknowledging the knowledge gap in the local
genetic landscape and the lack of a comprehensive
service model for patients with IEDs in Hong Kong,
we conducted this retrospective review to analyse
the local mutational spectrum across various IED
subtypes and the corresponding diagnostic yield in
our institution. Our aim was to better understand the
clinical utility of genomic testing in IED patients and
to formulate a comprehensive ocular genetic service
model that addresses the needs of local patients.
Methods
Study design and population
Patients presenting with eye manifestations were
retrospectively identified by querying records
between 1 December 2021 and 30 October 2023
through the Hospital Authority Teams database
under the CGSU/DCG at HKCH. The database
included all patients who had attended genetic
counselling clinics under the CGSU/DCG. Clinical
geneticists and ophthalmologists reviewed all
clinical notes, genetic reports, and electronic health
records in the Clinical Management System, as well
as paper records.
Patients’ phenotypes were reviewed and
categorised by ophthalmologists into the following
nine groups: (a) anterior segment dysgenesis; (b) IRDs; (c) cataract and lens disorders; (d)
microphthalmia, anophthalmia, and coloboma
spectrum; (e) neuro-ophthalmology (eg, optic
atrophy); (f) ocular albinism or oculocutaneous
albinism; (g) high myopia; (h) ocular tumours; and
(i) others.
Patients with inconclusive eye phenotypes were
excluded. Relevant history (including consanguinity,
ethnicity, and family history), physical examination
findings (dysmorphism and involvement of other
systems), ophthalmological assessments and
examinations, other relevant investigations (eg,
magnetic resonance imaging of the brain and renal
imaging), and previous genetic test reports were
reviewed. A positive family history was defined as
the presence of related eye phenotypes in a first-degree
relative, or in two or more second- or third-degree
relatives with the same condition.
All patients underwent comprehensive
dysmorphology evaluations and genetic counselling,
including pre-test and post-test consultations
with the clinical genetics team. Prior to providing
informed consent for genomic testing, patients were
counselled on the indications, limitations, diagnostic
yield, variants of uncertain clinical significance,
and the ethical, social, and legal implications of
genomic testing. Informed consent was obtained
from affected patients or their legal guardians before
undergoing diagnostic genomic testing.
Genomic testing
According to clinical indications, patients were
offered various genomic tests, including single-gene
sequencing, array comparative genomic
hybridisation, multiplex ligation–dependent
probe amplification, whole-exome sequencing–based panels, medical exome sequencing, and
mitochondrial sequencing. DNA was extracted
from peripheral blood ethylenediaminetetraacetic
acid samples. For mitochondrial sequencing,
mitochondrial DNA extracted from urine-derived
cells was used. All tests were performed in one of
two accredited laboratories: the Genetic Laboratory
of DH (which became a combined service with the
Hospital Authority after July 2023), or the Genetics
and Genomics Laboratory at HKCH, in accordance
with laboratory-specific protocols and guidelines.
Inheritance and phasing were determined via
targeted Sanger sequencing of parental samples.
Data collection and analysis
Clinical characteristics were collected from
electronic records and, when available, hospital case
notes and CGS paper records. These characteristics
included age at onset, age at first encounter,
sex, ethnicity, consanguinity, laterality of ocular
involvement, severity of visual impairment, family history of ocular conditions, syndromic features,
and other associated system involvement. Genetic
testing results were retrieved from the Clinical
Management System, CGS database, and paper
records. Additionally, reproductive planning (for
either the index patient or their parents) and other
subspecialty referrals after a substantiated molecular
diagnosis—as documented in genetic counselling
notes—were recorded for clinical utility analysis. All
clinical data are presented as percentages or means ± standard deviations, unless otherwise specified.
Molecular and clinical data from all recruited
individuals were analysed using SPSS (Windows
version 26.0; IBM Corp, Armonk [NY], United
States). Categorical variables (eg, syndromic vs
non-syndromic presentation, presence of family
history) were compared using Fisher’s exact test,
while continuous variables were compared using
the independent samples t test. P values of less than
0.05 were considered statistically significant. This
article was written in compliance with the STROBE
(Strengthening the Reporting of Observational
Studies in Epidemiology) reporting guidelines.
Results
Between December 2021 and October 2023, 3653
patients were registered at the HKCH genetic
counselling clinic. Of these, 148 symptomatic patients
from 147 families met the inclusion criteria for this
study. Approximately 4% of patients presented to
the genetics clinic with ophthalmological diseases.
Overall, 130 (87.8%) patients consented to genomic
testing (Fig).
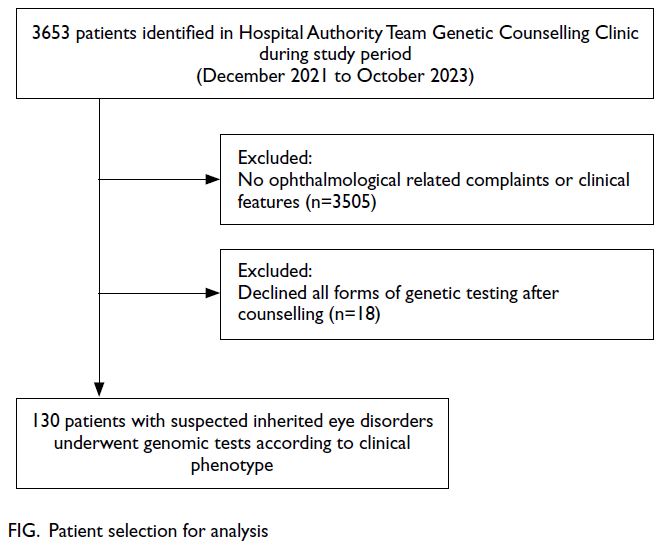
Figure. Patient selection for analysis
Patient demographics
Among the 130 patients, approximately 92% were
Chinese, with a male-to-female ratio of 3:2. The
mean age (±standard deviation) at onset was
12.5±16.2 years. Within this cohort, 53.1% of
patients were classified under IRDs, 14.6% under
neuro-ophthalmology, and 13.8% under cataract/lens disorders.
Fifteen patients (11.5% of those tested)
presented with more than one ocular phenotype.
The majority of patients (>80%) exhibited bilateral
ocular involvement. Detailed demographics, family
history, and disease categories of the 130 patients
who underwent genetic testing are presented in
Table 1.
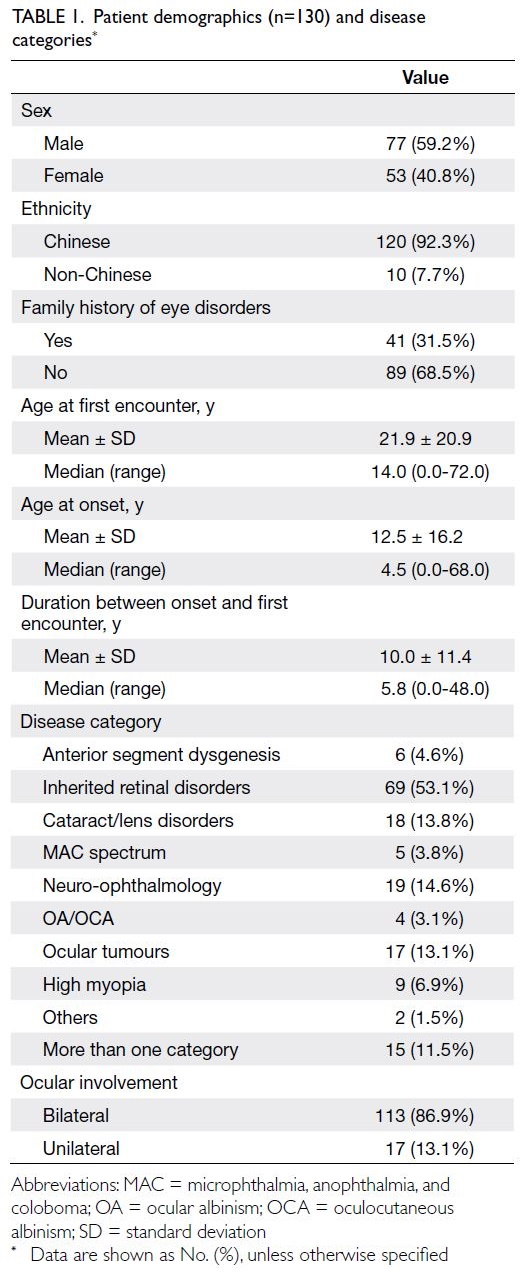
Table 1. Patient demographics (n=130) and disease categories
Molecular findings and diagnostic yield
The diagnostic yield of genomic testing was defined
as the proportion of individuals with pathogenic or
likely pathogenic molecular variants or structural
variants contributing to the clinical phenotype. A
whole-exome sequencing–based virtual panel was
requested for 78 (60%) of the 130 patients, based on
their presenting phenotypes (online supplementary Table 1). Using this panel-based approach, the
diagnostic yield was 51.3%. Twenty-three patients
(17.7%) underwent single-gene testing based on
highly specific phenotypes without molecular
heterogeneity, such as RB1, CHD7, NF1, and RS1
(online supplementary Table 2). This single-gene
approach successfully diagnosed 14 patients (60.8%).
Medical exome sequencing was offered to 22 patients
with multiple congenital anomalies or suspected
syndromes, achieving a diagnostic yield of 50%
(11/22). Two patients were diagnosed through copy
number variation analysis (online supplementary Table 2).
The overall diagnostic yield for this cohort
was 51.5% (Table 2). As mentioned earlier, 15
patients exhibited overlapping phenotypes across
disease categories, with inherited retinal disorders
and cataracts being the most common co-existing
phenotypes. The microphthalmia, anophthalmia,
and coloboma spectrum demonstrated the highest
diagnostic yield at 100%. All five patients in this
category presented with bilateral eye involvement
and were syndromic (eg, two with CHARGE
syndrome) [online supplementary Table 2]. Among
the 69 IRD patients who underwent testing, 40
had confirmed molecular diagnoses, resulting in
diagnostic yield of 58% for the IRD group. The most
commonly identified genes were USH2A, ABCA4,
COL2A1, RP1L1, and RS1 (online supplementary Table 2). No significant differences in diagnostic
yield were detected across disease categories (Table 2).
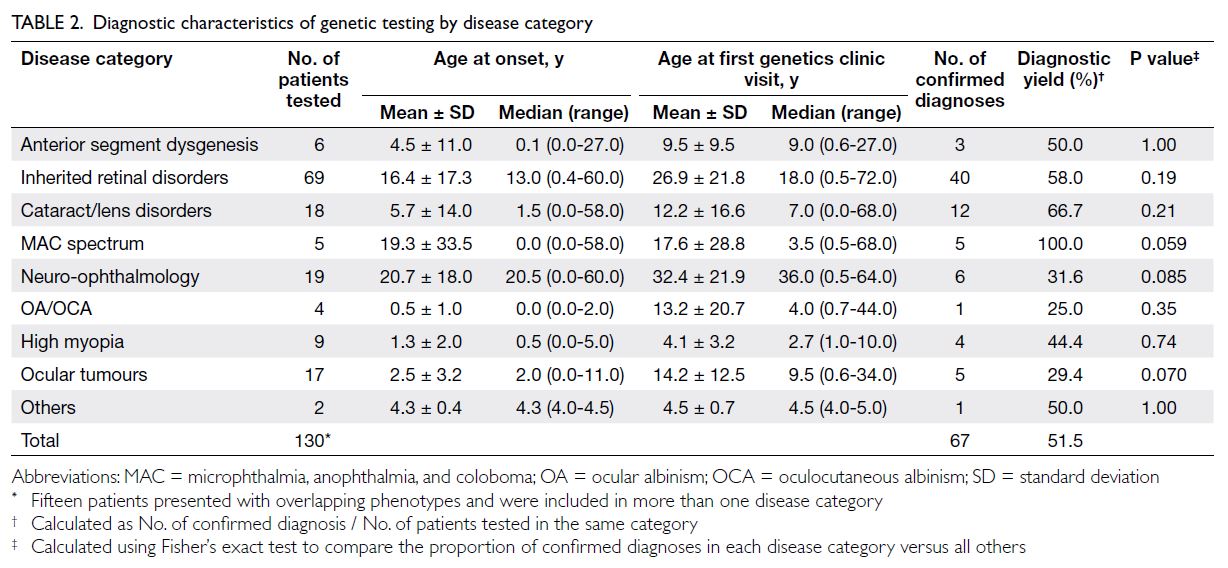
Table 2. Diagnostic characteristics of genetic testing by disease category
Patients presenting with IRDs and neuro-ophthalmological
conditions generally exhibited a later age at onset and age at first encounter compared
with other categories, although these differences
were not statistically significant (Table 2).
In total, 25 novel variants were identified in 25
patients across 20 genes. Of these, four remained of
uncertain clinical significance despite further phasing
and segregation analysis (online supplementary Table 2). Five variants were found in trans with
another likely pathogenic variant in the same gene, consistent with autosomal recessive inheritance.8
Following detailed phenotypic correlation and
variant curation, 16 previously unreported novel
variants were confirmed to contribute to molecular
diagnoses within this cohort.
A significant difference in the proportion of
positive genetic test results was observed between
patients with and without a family history of ocular
conditions (P=0.037). However, among patients
with a family history, the interval between symptom
onset and the first visit to the genetics clinic was
significantly longer. Positive molecular diagnoses
were also more likely to be achieved in syndromic
patients (P=0.0014) [Table 3].
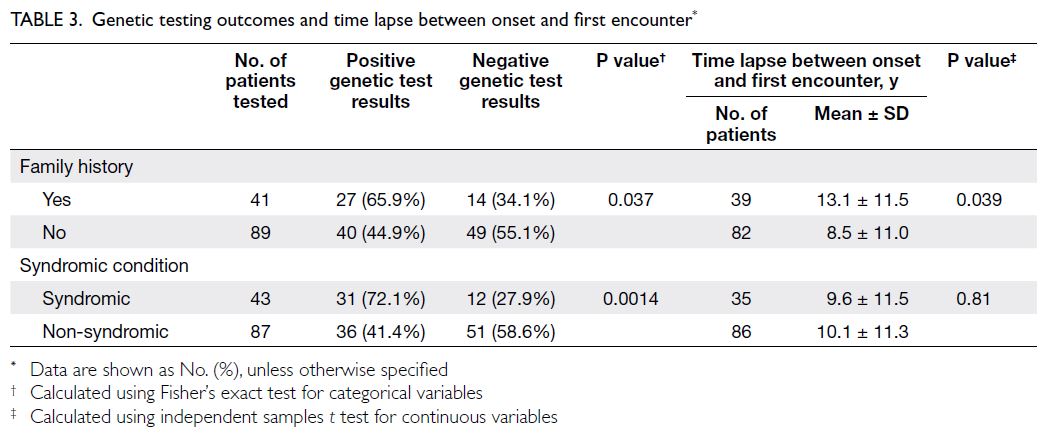
Table 3. Genetic testing outcomes and time lapse between onset and first encounter
As shown in Table 4, individuals with bilateral
eye involvement had a greater proportion of positive genetic test results (54.9%), although this
difference was not statistically significant (P=0.07).
Additionally, no significant difference in diagnostic
yield was observed according to age at onset (P=0.29).
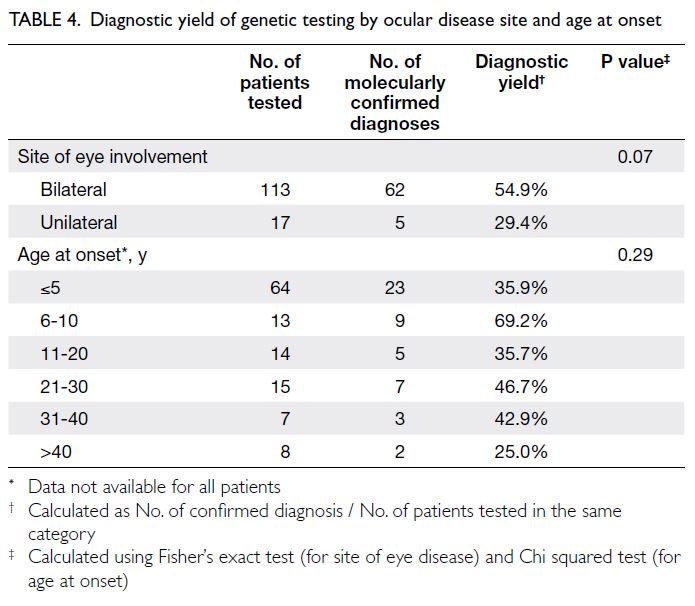
Table 4. Diagnostic yield of genetic testing by ocular disease site and age at onset
Diagnostic and clinical utilities
Genomic testing is increasingly recognised as an
important tool for establishing new diagnoses or
confirming ones, particularly in the context of rare
conditions, which are often complex and costly to
diagnose, leading to prolonged diagnostic odysseys.
Molecular findings may offer additional clinical
utility, including: (1) avoidance of unnecessary
investigations or treatments; (2) improved prognostic
certainty or redirection of clinical care; (3) enhanced
surveillance or timely referral for extraocular manifestations; (4) provision of pre-symptomatic
or cascade testing for potentially affected family
members; and (5) support for reproductive planning.
In total, 14 patients received revised
diagnoses after genomic testing, representing 21%
of positive cases (Table 5). These new diagnoses were related to syndromic conditions, such as
CTNNB1-related neurodevelopmental disorders, or
involved extraocular features, such as pantothenate
kinase–associated neurodegeneration (online supplementary Table 2).
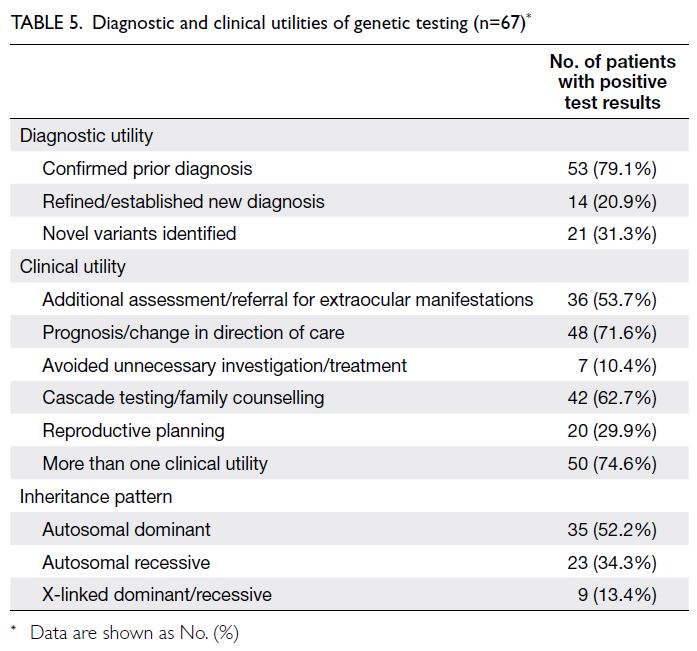
Table 5. Diagnostic and clinical utilities of genetic testing (n=67)
Through medical record review, we
determined that approximately 10% of test-positive
patients were able to avoid unnecessary
investigations and treatments. In two cases,
metabolic workups for congenital cataract were
discontinued after diagnostic confirmation. One
patient with a pathogenic ABCA4 variant was
advised to withhold vitamin A supplementation.
In another case, a syndromic diagnosis of SOX2-related microphthalmia eliminated the need for
repeated magnetic resonance imaging of the brain
and prompted clinicians to monitor for other
potential systemic associations, enabling timely
intervention. Overall, 74.6% of patients experienced
at least one clinical benefit as a result of genomic
testing. More than 70% of test-positive patients
benefited from improved prognostic certainty or a
redirection of care. Approximately 30% of patients—or their carrier or affected parents—were offered
options for reproductive planning through either
prenatal confirmatory testing or preimplantation
genetic testing. Table 5 summarises the clinical and
diagnostic utilities observed in this study.
Discussion
Molecular findings and diagnostic yield
In this cohort, we reviewed 130 patients who
attended the HKCH genetic counselling clinic over
a 23-month period. This review offers a snapshot
of the local genomic landscape of IEDs. The overall
diagnostic yield of molecular testing was 51.5%,
which is comparable to previously reported yields,
ranging from 25% to 70% depending on phenotype
and testing methodology.6 9 10 11 12 13 14 15 16 17 18 19 20
Among IRDs, a highly heterogeneous group,
the diagnostic yield was 58%. This finding is consistent
with a recent systematic review which reported a yield
of 61.3% (95% confidence interval=57.8%-64.7%)
across 51 studies of mixed IRD phenotypes.21 Several
studies have demonstrated that well-curated gene
panels are as effective as medical exome sequencing
in detecting pathogenic variants in patients with
IRDs.16 19 20 21 22
In our cohort of ocular tumours, 29.4% of
patients received germline molecular diagnoses;
most of these patients had unilateral retinoblastoma
with no family history. Neither routine next-generation
sequencing nor Sanger sequencing is
typically capable of detecting low-level mosaicism.
A previous study reported germline RB1 mutation
detection rates ranging from 10% to 55% in unilateral
retinoblastoma, which are substantially lower than those observed in bilateral cases.23 In the present
study, the oculocutaneous albinism/ocular albinism
group had the lowest diagnostic yield at 25%. This
low yield may be attributed to the small sample
size and the predominance of ocular albinism
cases, for which previous research has shown a
considerably lower molecular diagnostic yield than
oculocutaneous albinism.24
Four recurrent variants were identified in this
cohort (online supplementary Table 2):
- NM_000350.3 (ABCA4): c.1804C>T, p.(Arg602Trp). This variant is present at a very low frequency in the Genome Aggregation Database25 (gnomAD v2.1.1: 11 in 250 870 alleles), with a predominance in East Asian populations (gnomAD v2.1.1: 5 in 18 364 alleles). The exact carrier risk in our locality requires further research.
- NM_000330.4 (RS1): c.214G>A, p.(Glu72Lys). A missense variant located in exon 4 of the RS1 gene. This variant is well documented in Chinese populations, where it accounts for 9.2% of variants in individuals with X-linked retinoschisis.26
- NM_178857.6 (RP1L1): c.133C>T, p.(Arg45Trp). This hotspot mutation, located in exon 2 of RP1L1, is associated with occult macular dystrophy. Although its allele frequency is not particularly enriched in the Chinese population, it has been mentioned in case reports.27 28
- NM_206933.3 (USH2A): c.5572+1G>A. A splice-site variant in intron 27 of the USH2A gene, which has been documented in the literature.29 It has a relatively high allele frequency in East Asians (gnomAD v2.1.1: 3 in 249 996 alleles; East Asian subset: 3 in 18 382 alleles).30
Another variant, NM_153638.4 (PANK2):
c.655G>A, p.(Gly219Ser), is a rare missense
variant absent from the general population. It
was detected in our local database and reported
in 2023.31 Neurodegeneration with brain iron
accumulation 1A (OMIM #234200) is caused by
biallelic pathogenic variants in PANK2. This rare
condition is characterised by early-onset retinal
degeneration or pigmented retinopathy, followed
by subtle neurological deficits such as tremor
and extrapyramidal symptoms. Both ocular and
neurological features follow a progressive course.
Notably, two unrelated patients in our database
carried the same PANK2 variant. A large, population-based
study is warranted to determine whether this
variant represents a founder mutation in our locality.
Diagnostic and clinical utilities
Genomic testing has advanced considerably over
the past decade. As next-generation sequencing
technologies (eg, whole-genome sequencing and
multi-omics analysis) become more prevalent,
diagnostic yields continue to improve.32 Given the availability of existing therapies, such as voretigene
neparvovec for RPE65-related diseases, clinical trials
are increasingly investigating gene-based therapies,
including gene replacement through viral vectors,
mutation suppression via small molecules, and splice
modulation using antisense oligonucleotides.33 34
In addition to ending the diagnostic odyssey, a
molecular diagnosis informs clinical management,
facilitates access to other clinical services, initiates
surveillance for extraocular manifestations, and
supports family planning.6 12 35
Disease prognostication is a key aspect of
clinical utility, most commonly reported in the
IRD group. For example, COL2A1-related Stickler
syndrome carries a high risk of retinal detachment,
which may be mitigated through prophylactic
cryotherapy or laser retinopexy.36 37 38 Among patients
with unilateral retinoblastoma, those harbouring
germline variants require closer surveillance of
the contralateral eye and enhanced vigilance for
the potential development of other cancers later in
life.39 40
Among the 67 test-positive patients, 35 were
diagnosed with autosomal dominant conditions
(Table 5), six of which were inherited from an affected
parent. Approximately one-third of positive findings
were attributed to autosomal recessive conditions,
with both parents identified as heterozygous carriers.
Nine patients had X-linked conditions; in nearly all
cases, the mothers were confirmed as heterozygous
carriers, except for two who declined genetic testing.
In this context, molecular diagnosis is clearly
beneficial for cascade screening and reproductive
planning. In practice, however, the extent to which
patients report these benefits is often influenced
by age and family circumstances within the study
cohort. As a result, direct comparisons of reported
utility across studies remain challenging.
Limitations and strengths
In Hong Kong, our genetic counselling department
serves as the major referral centre, receiving patients
from both public and private sectors. Individuals
with more severe phenotypes are more likely to be
referred, resulting in potential ascertainment bias.
Genomic testing was recommended by
clinical geneticists based on the clinical phenotype.
However, due to resource limitations, not all patients
underwent the full spectrum of available tests,
which may have resulted in an underestimation of
the diagnostic yield. Additionally, certain clinical
subgroups (eg, microphthalmia, anophthalmia, and
coloboma) had limited sample sizes, potentially
affecting diagnostic yield outcomes. Despite these
limitations, this pilot study provides a reliable
estimate of the mutational spectrum and diagnostic
yield among local IED patients.
To our knowledge, this is the first retrospective study of IED patients to examine both the local
genetic landscape and the clinical utility of genomic
testing. Our findings highlight the importance of
integrating modern genomic technologies into
the management of patients with IEDs. They also
underscore the need for an enhanced service
model through a multidisciplinary team approach,
implemented via a combined ocular genetics clinic.
Ideally, clinical utility should be assessed
through a randomised controlled trial, which
maximises internal validity and control for
confounding variables. However, the level of evidence
required varies according to clinical indication
and type of genetic test. The data presented in this
retrospective observational study, collected over
nearly 2 years, are considered representative of real-world
clinical scenarios. Future research involving
multicentre collaborations over a longer period
(eg, 10 years) will provide a more comprehensive
understanding.
Ocular genetics clinic: a new service model in
Hong Kong
Interestingly, patients with a family history
experienced a longer interval between symptom
onset and their first encounter at the clinical
genetics clinic (Table 3). This finding emphasises the
importance of raising public awareness about the
role of genomic medicine in managing IEDs.
The multidisciplinary team clinic model—comprising ophthalmologists, genetic counsellors,
geneticists, and genetic nurses—is a current global
trend for integrating genomic testing into clinical care
pathways. It has been proven effective, particularly
when applied to IRDs as a model.41 42 Similar models
have also been adopted in other specialties, such as
neurogenetics and cardiogenetics clinics.
At HKCH, a combined ocular genetics clinic
commenced service in May 2022. The team includes
ophthalmologists, genetic counsellors, clinical
geneticists, optometrists, and nurses. Patients
are referred from both public and private sectors
for a variety of indications, such as atypical eye
phenotypes, suspected syndromic conditions, or
complex counselling needs (eg, variants of uncertain
clinical significance detected in previous genomic
tests conducted locally or overseas). This one-stop
combined clinic enables joint discussions among
specialists to formulate comprehensive management
plans and reduces the need for repeated hospital
visits, saving patients valuable time.
Conclusion
Approximately 4% of patients attending our genetic
clinic had ocular disorders. The overall diagnostic
yield of genomic testing was 51.5%; predominance
was the strongest among patients with syndromic
presentations and positive family history.
This study demonstrates high clinical utility
of genomic testing in over 70% of patients with
confirmed molecular diagnoses. There is a global
shift towards managing IED patients through a
multidisciplinary team clinic service model. To
meet the growing demand for genomic medicine in
IEDs, future studies should incorporate prospective,
population-wide sampling, long-term follow-up,
and multicentre collaboration.
Author contributions
Concept or design: SSW Cheng, HM Luk.
Acquisition of data: SSW Cheng.
Analysis or interpretation of data: SSW Cheng, SSL Cheung.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: SSW Cheng.
Analysis or interpretation of data: SSW Cheng, SSL Cheung.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
As an editor of the journal, JCS Yam was not involved in the peer review process. Other authors have disclosed no conflicts
of interest.
Acknowledgement
The authors thank the patients and their families for contributing the clinical data used in this study.
Declaration
Part of the research data was presented at Kowloon Central Cluster Convention, 25 October 2024, Hong Kong.
Funding/support
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
This research was performed in compliance with the
Declaration of Helsinki. Ethics approval was granted by the
Hospital Authority Central Institutional Review Board, Hong
Kong (Ref No.: PAED-2023-076). A waiver of patient consent
was obtained from the Committee due to the retrospective
nature of the research.
Supplementary material
The supplementary material was provided by the authors and
some information may not have been peer reviewed. Accepted
supplementary material will be published as submitted by the
authors, without any editing or formatting. Any opinions
or recommendations discussed are solely those of the
author(s) and are not endorsed by the Hong Kong Academy
of Medicine and the Hong Kong Medical Association.
The Hong Kong Academy of Medicine and the Hong Kong
Medical Association disclaim all liability and responsibility
arising from any reliance placed on the content.
References
1. National Academies of Sciences, Engineering, and
Medicine; Health and Medicine Division; Board on Health
Care Services; Board on the Health of Select Populations;
Committee on the Evidence Base for Genetic Testing. An
Evidence Framework for Genetic Testing. Washington
(DC): National Academies Press (US); 2017.
2. Rahi JS, Cable N; British Childhood Visual Impairment
Study Group. Severe visual impairment and blindness in
children in the UK. Lancet 2003;362:1359-65. Crossref
3. Chen HY, Lehmann OJ, Swaroop A. Genetics and therapy
for pediatric eye diseases. EBioMedicine 2021;67:103360. Crossref
4. FDA approves hereditary blindness gene therapy. Nat
Biotechnol 2018;36:6. Crossref
5. Stone EM, Aldave AJ, Drack AV, et al. Recommendations
for genetic testing of inherited eye diseases: report of
the American Academy of Ophthalmology task force on
genetic testing. Ophthalmology 2012;119:2408-10. Crossref
6. Burdon KP. The utility of genomic testing in the
ophthalmology clinic: a review. Clin Exp Ophthalmol
2021;49:615-25. Crossref
7. Lam ST, To CH, Leung KW, Yip SP, Lo IF, Tsang KP. Lessons
learnt from a genetic disease registry in Hong Kong. Hong
Kong Med J 2021;27:226-8. Crossref
8. Richards S, Aziz N, Bale S, et al. Standards and guidelines
for the interpretation of sequence variants: a joint
consensus recommendation of the American College of
Medical Genetics and Genomics and the Association for
Molecular Pathology. Genet Med 2015;17:405-24. Crossref
9. Ma A, Grigg JR, Flaherty M, et al. Genome sequencing in
congenital cataracts improves diagnostic yield. Hum Mutat
2021;42:1173-83. Crossref
10. Haug P, Koller S, Maggi J, et al. Whole exome sequencing
in coloboma/microphthalmia: identification of novel
and recurrent variants in seven genes. Genes (Basel)
2021;12:65. Crossref
11. García Bohórquez B, Aller E, Rodríguez Muñoz A, Jaijo T,
García García G, Millán JM. Updating the genetic
landscape of inherited retinal dystrophies. Front Cell Dev
Biol 2021;9:645600. Crossref
12. Lenassi E, Clayton-Smith J, Douzgou S, et al. Clinical utility
of genetic testing in 201 preschool children with inherited
eye disorders. Genet Med 2020;22:745-51. Crossref
13. Wang P, Li S, Sun W, et al. An ophthalmic targeted exome
sequencing panel as a powerful tool to identify causative
mutations in patients suspected of hereditary eye diseases.
Transl Vis Sci Technol 2019;8:21. Crossref
14. Patel A, Hayward JD, Tailor V, et al. The oculome panel test:
next-generation sequencing to diagnose a diverse range
of genetic developmental eye disorders. Ophthalmology
2019;126:888-907. Crossref
15. Martin-Merida I, Avila-Fernandez A, Del Pozo-Valero M,
et al. Genomic landscape of sporadic retinitis pigmentosa:
findings from 877 Spanish cases. Ophthalmology
2019;126:1181-8. Crossref
16. Wang L, Zhang J, Chen N, et al. Application of whole
exome and targeted panel sequencing in the clinical
molecular diagnosis of 319 Chinese families with inherited
retinal dystrophy and comparison study. Genes (Basel)
2018;9:360. Crossref
17. Lasseaux E, Plaisant C, Michaud V, et al. Molecular
characterization of a series of 990 index patients with
albinism. Pigment Cell Melanoma Res 2018;31:466-74. Crossref
18. Haer-Wigman L, van Zelst-Stams WA, Pfundt R, et al.
Diagnostic exome sequencing in 266 Dutch patients with
visual impairment. Eur J Hum Genet 2017;25:591-9. Crossref
19. Saudi Mendeliome Group. Comprehensive gene panels
provide advantages over clinical exome sequencing for
Mendelian diseases. Genome Biol 2015;16:134. Crossref
20. Consugar MB, Navarro-Gomez D, Place EM, et al. Panel-based
genetic diagnostic testing for inherited eye diseases
is highly accurate and reproducible, and more sensitive
for variant detection, than exome sequencing. Genet Med
2015;17:253-61. Crossref
21. Britten-Jones AC, Gocuk SA, Goh KL, Huq A, Edwards TL,
Ayton LN. The diagnostic yield of next generation
sequencing in inherited retinal diseases: a systematic
review and meta-analysis. Am J Ophthalmol 2023;249:57-73. Crossref
22. Hayman T, Millo T, Hendler K, et al. Whole exome
sequencing of 491 individuals with inherited retinal diseases
reveals a large spectrum of variants and identification of
novel candidate genes. J Med Genet 2024;61:224-31. Crossref
23. Gupta H, Malaichamy S, Mallipatna A, et al. Retinoblastoma
genetics screening and clinical management. BMC Med
Genomics 2021;14:188. Crossref
24. Chan KS, Bohnsack BL, Ing A, et al. Diagnostic yield of
genetic testing for ocular and oculocutaneous albinism in
a diverse united states pediatric population. Genes (Basel)
2023;14:135. Crossref
25. Genome Aggregation Database. Available from: https://gnomad.broadinstitute.org/. Accessed 1 Sep 2024.
26. Huang L, Sun L, Wang Z, et al. Clinical manifestation
and genetic analysis in Chinese early onset X-linked
retinoschisis. Mol Genet Genomic Med 2020;8:e1421. Crossref
27. Qi YH, Gao FJ, Hu FY, et al. Next-generation sequencing–aided rapid molecular diagnosis of occult macular
dystrophy in a Chinese family. Front Genet 2017;8:107. Crossref
28. Xiao S, Sun W, Xiao X, et al. Clinical and genetic features
of retinoschisis in 120 families with RS1 mutations. Br J
Ophthalmol 2023;107:367-72. Crossref
29. Lin YW, Huang YS, Lin CY, et al. High prevalence of
exon-13 variants in USH2A-related retinal dystrophies in
Taiwanese population. Orphanet J Rare Dis 2024;19:238. Crossref
30. Chau JF, Yu MH, Chui MM, et al. Comprehensive analysis
of recessive carrier status using exome and genome
sequencing data in 1543 Southern Chinese. NPJ Genom
Med 2022;7:23. Crossref
31. Wong EW, Cheng SS, Woo TT, Lam RF, Lai FH. Concurrent
PANK2 and OCA2 variants in a patient with retinal
dystrophy, hypopigmented irides and neurodegeneration.
Ophthalmic Genet 2023;44:403-7. Crossref
32. Weisschuh N, Mazzola P, Zuleger T, et al. Diagnostic
genome sequencing improves diagnostic yield: a
prospective single-centre study in 1000 patients with
inherited eye diseases. J Med Genet 2024;61:186-95. Crossref
33. Drag S, Dotiwala F, Upadhyay AK. Gene therapy for retinal
degenerative diseases: progress, challenges, and future
directions. Invest Ophthalmol Vis Sci 2023;64:39. Crossref
34. Tan F, Li X, Wang Z, Li J, Shahzad K, Zheng J. Clinical
applications of stem cell–derived exosomes. Signal
Transduct Target Ther 2024;9:17. Crossref
35. Sahu A, Kaur S, Sukhija J, Srivastava P, Kaur A. Spectrum
of congenital and inherited ocular disorders seen in a
genetic clinic: experience of a developing ocular genetic
service. Indian J Ophthalmol 2023;71:935-40. Crossref
36. Fincham GS, Pasea L, Carroll C, et al. Prevention of
retinal detachment in Stickler syndrome: the Cambridge
prophylactic cryotherapy protocol. Ophthalmology
2014;121:1588-97. Crossref
37. Savarirayan R, Bompadre V, Bober MB, et al. Best practice
guidelines regarding diagnosis and management of patients
with type II collagen disorders. Genet Med 2019;21:2070-80. Crossref
38. Khanna S, Rodriguez SH, Blair MA, Wroblewski K,
Shapiro MJ, Blair MP. Laser prophylaxis in patients with
Stickler syndrome. Ophthalmol Retina 2022;6:263-7. Crossref
39. Tonorezos ES, Friedman DN, Barnea D, et al.
Recommendations for long-term follow-up of adults with heritable retinoblastoma. Ophthalmology 2020;127:1549-57.Crossref
40. Abramson DH. Re: Skalet et al.: Screening children at risk
for retinoblastoma: consensus report from the American
Association of Ophthalmic Oncologists and Pathologists
(Ophthalmology. 2018;125:453-458). Ophthalmology
2018;125:e63-4. Crossref
41. Davison N, Payne K, Eden M, et al. Exploring the
feasibility of delivering standardized genomic care using
ophthalmology as an example. Genet Med 2017;19:1032-9. Crossref
42. Black GC, Sergouniotis P, Sodi A, et al. The need for widely
available genomic testing in rare eye diseases: an ERN-EYE
position statement. Orphanet J Rare Dis 2021;16:142. Crossref