Hong Kong Med J 2025;31:Epub 5 Jun 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Fragile X syndrome: genetic and clinical profile in the Hong Kong Chinese population
Candice WM Au, MB, BS, FHKAM (Paediatrics)1; HM Luk, MD, FHKAM (Paediatrics)1; Stephanie Ho, MB, ChB, FHKAM (Paediatrics)1; SW Cheng, MB, ChB, FHKAM (Paediatrics)1; Stephen TS Lam, MD, FHKAM (Paediatrics)2; Brian HY Chung, MD, FHKAM (Paediatrics)3; SC Chong, MB, BS, FHKAM (Paediatrics)4; Ivan FM Lo, MB, ChB, FHKAM (Paediatrics)1
1 Department of Clinical Genetics, Hong Kong Children’s Hospital, Hong Kong SAR, China
2 Clinical Genetics Service, The Hong Kong Sanatorium & Hospital, Hong Kong SAR, China
3 Department of Paediatrics and Adolescent Medicine, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China
4 Department of Paediatrics, The Chinese University of Hong Kong, Hong Kong SAR, China
Corresponding author: Dr Ivan FM Lo (dr.ivanlo@gmail.com)
 Full paper in PDF
Full paper in PDF
Abstract
Introduction: Fragile X syndrome (FXS) is a
common inherited cause of intellectual disability,
and FXS testing is recommended as a first-line
genetic investigation for global developmental delay
or intellectual disability. This retrospective study
evaluated the diagnostic yield of FXS testing and
clinical features in Chinese patients in Hong Kong.
Methods: From 1993 to 2022, 7291 patients
referred to the Clinical Genetic Service for
neurodevelopmental conditions (eg, developmental
delay, autism spectrum disorder, and intellectual
disability) underwent FXS testing. In total, 103
individuals from 61 families were confirmed to
have an FMR1 full mutation, including 59 index
cases and 44 family members. Clinical features of
70 Chinese patients with FXS, including growth,
neurobehavioural features, and other co-morbidities,
were evaluated.
Results: The diagnostic yield of FXS testing was
0.8%. The median age at diagnosis for index cases
was 4.1 years, with a trend towards earlier diagnosis
in recent years. In 27 families (44.2%), multiple
members carried a full mutation. Prenatal diagnosis
was arranged in 11% of families. Developmental
delay was observed in all males, compared with
45.0% of females. Intellectual disability affected
86.0% of males but only 30.0% of females. Common
co-morbidities included obesity, autism spectrum
disorder, attention-deficit/hyperactivity disorder, epilepsy, gastrointestinal problems, and sleep
disturbances. Features such as strabismus, scoliosis,
and mitral valve prolapse were rarely reported.
Conclusion: Fragile X syndrome is more than a
pure neurodevelopmental disorder. Our findings
highlight the importance of early diagnosis and
subsequent management, with awareness of relevant
surveillance and management guidelines.
New knowledge added by this study
- The local diagnostic yield of fragile X syndrome in patients referred for developmental delay/intellectual disability is 0.8%. There is a temporal trend towards earlier diagnosis. This study explored the landscape of cascade screening and prenatal diagnosis in Hong Kong.
- We examined the co-morbidity profile of patients with a full mutation in the FMR1 gene in Hong Kong. We observed a substantial number of co-morbidities beyond neurodevelopmental issues, requiring regular follow-up and surveillance.
- There is a need for heightened awareness of disease-specific surveillance guidelines, which may be facilitated by the development of rare disease registries.
- Integration of structured surveillance protocols into routine care for patients with fragile X syndrome may improve early identification and management of co-morbidities, thereby enhancing long-term health outcomes.
Introduction
Fragile X syndrome (FXS; OMIM #300624), an
X-linked dominant condition, is one of the most
common inherited causes of intellectual disability (ID)1 2 3 and autism spectrum disorder (ASD).2 3 4 5 The
prevalence of FXS is most widely regarded as 1 in
4000 for males and 1 in 8000 for females.6 7 8 9 Fragile
X syndrome is within the spectrum of FMR1-related disorders,10 caused by pathogenic variants in the FMR1 (fragile X messenger ribonucleoprotein 1)
gene (OMIM #309550) mapped to the chromosome
Xq27.3 region, which encodes the fragile X mental
retardation protein.
Fragile X syndrome is the first genetic
disorder known to be caused by trinucleotide repeat
expansions—specifically, cytosine-guanine-guanine
(CGG) repeats in the 5’ untranslated region of the
FMR1 gene. FMR1 alleles are categorised as normal
(<45), intermediate (45-54), premutation (PM,
55-200), and full mutation (FM, >200) based on
repeat size. Premutation alleles are associated with
elevated levels of FMR1 messenger ribonucleic acid,10
leading to ribonucleic acid toxicity that can result
in fragile X–associated tremor/ataxia syndrome,
fragile X–associated primary ovarian insufficiency,
or fragile X–associated neuropsychiatric disorders.10
Conversely, FXS typically results from FM with
promoter region hypermethylation and histone
protein deacetylation,11 12 causing transcriptional
silencing.13 14 Most individuals inherit the FM from
their mothers, who are PM carriers. Stability upon
maternal transmission depends on the size of the
PM.15
Characteristic signs of FXS, including
prominent ears, elongated face, protruding ears,
and macroorchidism, tend to evolve with age.1 4 Facial dysmorphism can vary depending on ethnic
background,4 and females exhibit greater clinical
variability.16 17 Most patients are not diagnosed until the age of 3 years.18 19 Fragile X syndrome is also
associated with multiple medical co-morbidities,
such as recurrent otitis media, mitral valve prolapse,
and connective tissue problems.3
Clinical presentation can be further
complicated by either size mosaicism or methylation
mosaicism.20 Size mosaicism refers to cell populations
with variably sized CGG repeats—typically the
presence of PM or intermediate/normal alleles in
addition to FMs. Methylation mosaicism involves
both methylated and unmethylated cell populations
at the FMR1 locus. Mosaicism in males with FXS has
been reported in 12% to 41% of cases.21 22 23
While the epidemiology of FXS has been
extensively studied in Western populations,6 7 8 9 the
reported prevalence of FXS among Chinese patients
with developmental delay or ID showed variability
(ranging from 0.43% to 12.9%).24 25 Furthermore,
the prevalence of medical co-morbidities remains
understudied in the Chinese population.
In this single-centre retrospective study, we
aimed to: (1) review the clinical features of FXS
patients referred to the Department of Clinical
Genetics of the Hospital Authority (formerly the
Clinical Genetic Service of the Department of Health);
(2) evaluate parameters regarding growth, medical
co-morbidities, and neurobehavioural features in the
Hong Kong Chinese patient population with FXS; (3)
assess the diagnostic yield of FXS testing in patients
with unexplained developmental delay or ID; and
(4) review the diagnostic journey of such patients.
Methods
Patient data
Neurodevelopmental delay, ID, or ASD are the
main reasons for ordering FXS testing. Over the
30-year period from 1993 to 2022, 7291 patients
referred for such neurodevelopmental conditions
underwent FXS molecular testing after clinical
genetic evaluation. Maternal testing and further
cascade testing were considered upon diagnosis in
index cases.
Patients with FMR1 FMs were included in the
initial analysis, and a retrospective chart review of
printed and electronic records was performed. For
analysis of clinical features among Chinese patients
with FXS, individuals who self-identified as non-Chinese or had co-existing copy number variants
or chromosomal structural abnormalities were
excluded.
Molecular data
Genomic DNA was extracted from peripheral blood leucocytes using standardised methods, in accordance with the manufacturer’s instructions.
Prior to 2014, polymerase chain reaction (PCR)
followed by Southern blot analysis was used to
identify individuals with FXS. This approach was
subsequently replaced by conventional PCR that can
detect (CGG)n alleles up to 90 repeats, followed by
triplet-primed PCR and methylation-specific PCR
using the AmplideX kit (Asuragen, Austin [TX], US),
if necessary.
Statistical analysis
Baseline demographic characteristics were
descriptively summarised. Continuous variables were
reported as means and standard errors for normally
distributed data, and as medians and ranges/interquartile ranges (IQRs) for non-parametrically
distributed data. To assess the association between
age at diagnosis and year of assessment, correlation
analysis was performed using the Pearson correlation
coefficient (r), with a statistical significance threshold
of 5%. Prevalence proportions were used to evaluate
categorical clinical characteristics. Comparisons
between males and females were made using the Chi
squared test or Fisher’s exact test. Statistical analysis
was performed using SPSS (Windows version 26.0; IBM Corp, Armonk [NY], US).
Results
Patient demographics
Overall, 103 individuals from 61 families were
confirmed to have an FM in the FMR1 gene. Index
cases were defined as patients referred from their
parent institution for their condition. Among the
index cases, eight individuals came from four families,
with two affected members referred separately in
each family. In six other families, the consultand
was an unaffected member referred due to a positive
family history. Family screening identified 44
additional cases in 29 families, comprising 13 males
(29.5%) and 31 females (70.5%) [Table 1].
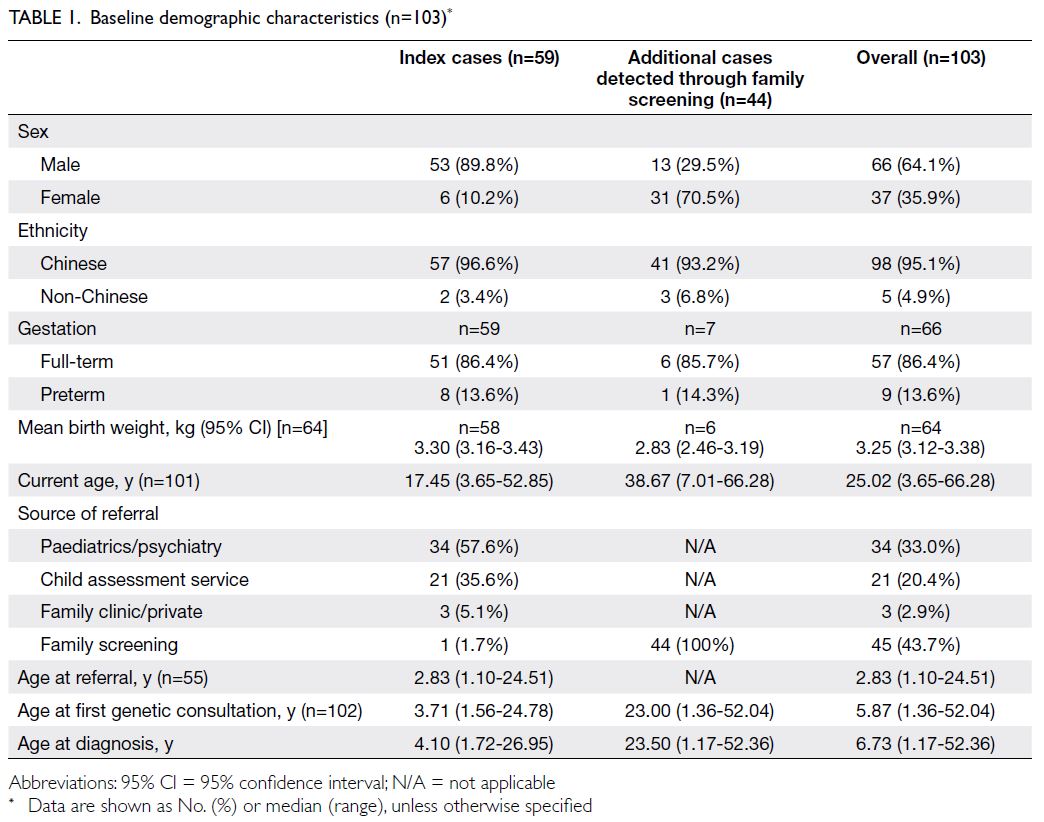
Table 1. Baseline demographic characteristics (n=103)
Family history
Details of family history for 55 unrelated index
cases and six consultands are presented in Table 2.
Overall, 41 (67.2%) had a positive family history in
one or more aspects.
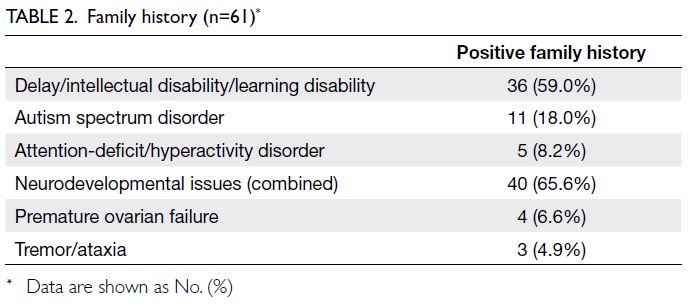
Table 2. Family history (n=61)
Diagnosis
Of 7291 patients underwent testing, 59 index cases were identified, yielding an overall diagnostic rate
of 0.8%. The sex-specific diagnostic yields were
1.0% for males and 0.3% for females. Additionally,
one male and one female patient had PMs. There
was an upward trend in the number of FXS tests
performed (unpublished data). The median ages at
diagnosis were 6.73 years (range, 1.17-52.36) among
all FXS patients (including those identified through
family screening) and 4.10 years (range, 1.72-26.95)
when considering index cases alone. The median
diagnostic lag time for index cases, defined as the
time elapsed between referral and diagnosis, was
11.0 months (IQR=6.53-20.0, n=54).
The temporal trends in diagnosis are shown
in Table 3. A weak negative correlation between
age and assessment year was observed for all cases
(r=-0.267, n=103; P=0.006). Regarding index cases,
a moderate negative correlation was observed
(r=-0.396, n=59; P=0.0019), suggesting a trend
towards earlier diagnosis over time.
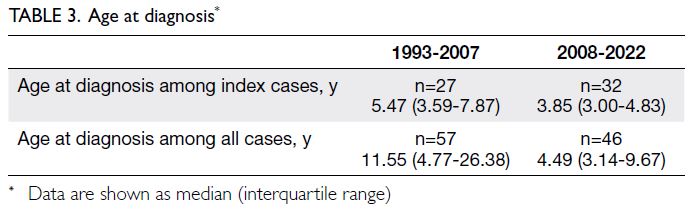
Table 3. Age at diagnosis
The mosaicism statuses of our patients are summarised in Table 4.

Table 4. Mosaicism status
Family cascade testing
Among the 61 families, 54 underwent maternal
testing—44 were PM carriers and 10 were FM
carriers. Cascade testing was conducted in other
family members in 45 families (73.8%). Twenty
siblings were identified as affected individuals, and
maternal second-/third-degree relatives constituted
another 13 cases. In 27 families (44.2%), more than
one FM carrier was identified—15 families (24.6%)
had two affected members, nine (14.8%) had three
affected members, and three (4.9%) had four affected
members. Nonetheless, 16 families (26.2%) did not
proceed with further cascade testing after maternal
testing. Four families (6.6%) did not undergo any
family testing at all.
Prenatal diagnosis was arranged for 11 families
(18%), involving 10 PM carriers and two FM carriers.
Two male fetuses were affected by FM, and these
pregnancies were terminated. One FM carrier opted
for termination of pregnancy at 10 weeks of gestation
despite counselling regarding the availability of
prenatal diagnosis.
Clinical features
Seventy Chinese patients with FMR1 FM from 55 different families were included in the analysis of clinical features (Fig); details are summarised in
Table 5.
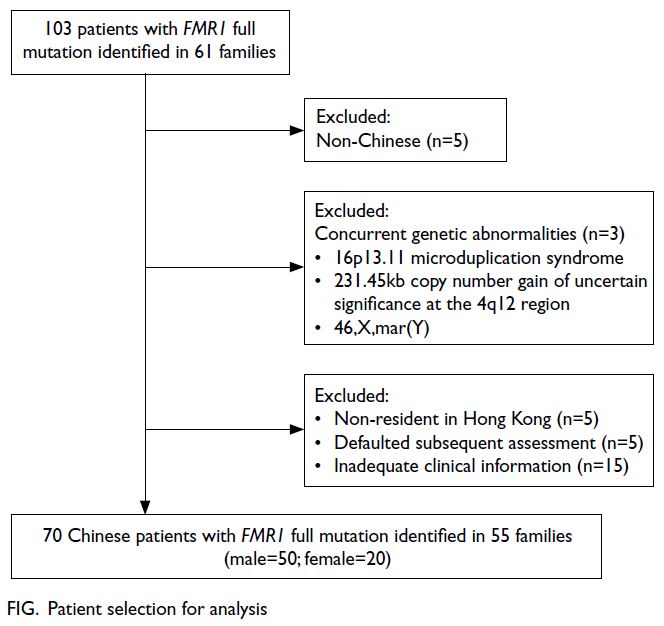
Figure. Patient selection for analysis
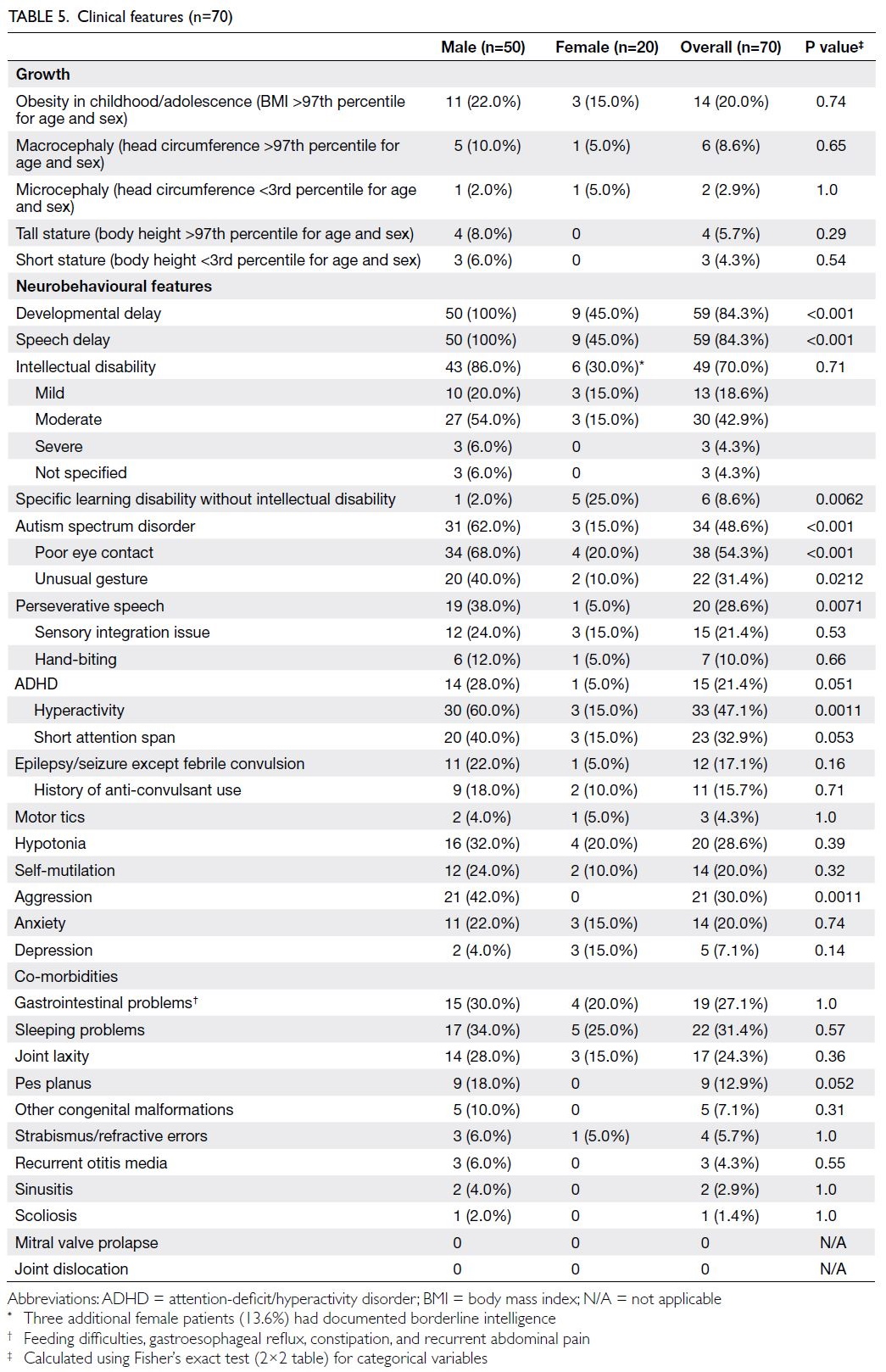
Table 5. Clinical features (n=70)
The presence and severity of ID, co-morbid
ASD, or attention-deficit/hyperactivity disorder
were determined based on clinician reports. More
than half of the male patients (54.0%) had ID of
moderate or greater severity. None of the female
patients had severe ID; three females had borderline
intelligence not supporting a diagnosis of ID.
Epilepsy was diagnosed in 12 patients (17.1%).
One 10-year-old boy with refractory epilepsy had
high-risk medulloblastoma and completed treatment
at age 6 years. He developed spasm-like attacks and
possible focal seizures at age 7 years. Among the
remaining patients, eight had generalised seizures,
two had a mixed semiology of generalised and
focal seizures, and one patient had unclear seizure
semiology. The age at seizure onset ranged from 2
to 19 years, with a median of 7.0 years (IQR=3.75-8.0). Three patients experienced convulsive status
epilepticus triggered by infective episodes, one
required intensive care unit admission.
Forty-three patients (61.4%) underwent
neuroimaging (magnetic resonance imaging/computed tomography of the brain), and most results were unremarkable.
Eight patients (five males and three females)
with mosaicism were eligible for analysis of clinical
features after excluding individuals with inadequate
data. These patients generally had less severe ID than
non-mosaic patients, although proper comparison
was hindered by the small sample size.
Gastrointestinal conditions and sleep
problems were common co-morbidities, affecting
27.1% and 31.4% of patients, respectively. Seven
patients underwent echocardiography at least
once; two displayed transient aortic root dilatation.
Congenital anomalies identified among our patients
included Pierre Robin sequence, Klippel-Trenaunay
syndrome, hemifacial asymmetry, microtia, and
pigmentary mosaicism. These conditions were
relatively rare in the literature.
Discussion
Clinical features
Approximately 20% of our patients developed obesity
in childhood or adolescence, which aligns with the
general childhood overweight/obesity prevalence in
Hong Kong (around 20%).26 However, a US study27
examining 848 families with at least one child had
FXS showed that 31% of male and 15% of female
children were obese. With respect to obesity alone,
the frequency may be higher among our patients than
in the general population, which may be attributed
to physical inactivity in individuals with ID, as well
as the use of psychiatric medications.
Five male patients (10.0%) and one female
patient (5.0%) exhibited macrocephaly, and a few had
suspected overgrowth syndrome upon referral. A
subset of FXS patients has been reported to present
with Sotos- or Prader-Willi–like phenotypes.16 This
feature may pose a diagnostic challenge.
In our study, the frequency of developmental
delay and ID was consistent with findings in
other populations. Female patients displayed
milder phenotypes, which is compatible with the
presentation of X-linked disorders. Additionally, 48.6% of patients had a clinician-reported diagnosis
of ASD. The reported prevalence of co-morbid ASD
in males with FXS varies widely across studies, from
30% to 60%.3 28 The use of different instruments has
been reported to cause diagnostic inconsistency;
this is further complicated by the intrinsic difficulty
in diagnosing ASD among individuals with ID. The
frequencies of hyperactivity and attention-deficit/hyperactivity disorder in our study are similar to
rates in the literature (50%-60% and 12%-23%,
respectively),29 but smaller percentages of our
patients displayed inattention, anxiety problems,
or depression compared to the literature (74%-84%
for inattention, 58%-86% for anxiety problems, and
8%-12% for depression).29 The lower rates of such
conditions in our study may be due to diagnostic
overshadowing. Active research is underway to
identify more accurate diagnostic measures for
neurobehavioural co-morbidities.28
Overall, 17.1% of our patients displayed
epilepsy, with a predilection towards generalised
seizures. This is in agreement with the work of Berry-Kravis et al,30 who characterised seizures in the largest evaluated cohort of FXS patients, although
earlier case series suggested that focal onset seizures
with impaired awareness were the most common
semiology.30 Notably, three patients presented with
convulsive status epilepticus, which is uncommon
among FXS patients.
The presence of co-morbidities such as
gastrointestinal problems, sleep disturbances, joint
laxity, and pes planus was consistent with commonly
observed clinical patterns in individuals with FXS.
Nonetheless, only a small percentage of patients in
our cohort showed strabismus or refractive errors,
scoliosis, or recurrent otitis media; none exhibited
joint dislocations or mitral valve prolapse (Table 5).
The true prevalence of mitral valve prolapse remains
unclear. Loehr et al31 reported a prevalence as high
as 55% in a series of FXS patients in 1986, whereas
Kidd et al3 reported a prevalence of 0.8%; some Asian
studies32 33 did not identify any individuals with
mitral valve prolapse.
A systematic approach to health supervision
for FXS has been recommended by the American
Academy of Pediatrics1 28 across developmental
stages. To our knowledge, there are no established
surveillance guidelines in Hong Kong. Ultimately,
FXS is more than a purely neurodevelopmental
disorder; it is important to be aware of potential
multisystemic approach and provide health
supervision as needed.
Diagnosis
Our diagnostic yield of 0.8% is consistent with a
local study in 1999,34 which showed a diagnostic
yield of 0.6% among 324 patients with mild ID of
unspecified cause, and with a study by Chen et al (0.93%)35 that evaluated the diagnostic yield of FXS
testing in 553 unrelated patients with moderate
to severe ID of unknown cause in Beijing in 2015.
Nonetheless, our yield is slightly lower than those
reported by Mei et al (2.4%)32 and Zhong et al (2.8%),36 which were derived from relatively large-scale
studies conducted in Chinese populations. Our
results also revealed a slightly lower diagnostic yield
compared with that of Western literature, which
is around 1.5% to 2%.37 This may be explained by
reported differences in the distribution of normal,
PM, and FM alleles between Asian and non-Asian
populations. Various studies have identified a lower
prevalence of PM alleles in East Asians compared
with Western populations. One study reported that
the prevalence of PM and asymptomatic FM carriers
in the Hong Kong Chinese pregnant population
was 1 in 883,38 whereas another study showed a
prevalence of 1 in 1113 among unaffected Chinese
individuals.39 The reported prevalence of PM alleles
in Western populations varies from 1 in 113 to 1
in 382, depending on ethnicity.39 Intriguingly, most
FMR1 alleles contain 29 or 30 CGG repeats across
different populations, including ours. Alternatively,
the apparent difference in PM allele prevalence may
be explained by the founder haplotype hypothesis,
whereby various factors contribute to disparate rates
of normal-to-PM transitions, including different
AGG interruption patterns across populations.40
Although preliminary studies have explored an
association between neurodevelopmental difficulties
and PM status, findings have been inconclusive.
In our cohort, only two patients referred for
developmental delay exhibited PM status.
Our study showed a weak but statistically
significant trend towards an earlier age at diagnosis,
which may be attributed to increased awareness of
children’s developmental needs and, consequently,
an earlier age at referral. The median age at diagnosis
was 4.1 years for index cases alone, and 6.73 years for
all cases in our study. These values are comparable to
international data where the average age at diagnosis
ranges from 2.9 to 6.3 years.18 33
There has been debate regarding whether FXS
testing should be utilised as a first-line investigation
to evaluate developmental delay. However, it is a
simple and inexpensive test with a short turnaround
time. The availability of such a test is crucial because
it aids in prompt diagnosis, facilitating further
cascade testing and reproductive planning. In our
study, 44.2% of families had more than one affected
member. Ten female PM carriers and two FM carriers
from 11 families (18%) underwent prenatal diagnosis;
two pregnancies were terminated after identification
of FXS status. A diagnosis in one family member
may influence others’ decisions regarding pregnancy
and subsequently affect pregnancy outcomes.
Fragile X PM carrier screening is recommended by organisations such as the American College of
Obstetricians and Gynecologists41 and the American
College of Medical Genetics and Genomics42 for
women with a family history suggestive of fragile
X—related disorders who are either considering
pregnancy or currently pregnant. Although prenatal
carrier testing is free for women of childbearing age
in some countries, it is currently self-financed in
Hong Kong and thus not widely implemented.
An expedited diagnosis can facilitate the
timely implementation of medical interventions. For
PM carriers who exhibit increased risks of fragile X—associated primary ovarian insufficiency and fragile
X—associated tremor/ataxia syndrome, anticipatory
guidance and timely referrals can be provided.
Furthermore, multiple targeted therapeutic agents
with the potential to reverse some neurobiological
aspects of the disorder (eg, mavoglurant, metformin,
cannabidiol transdermal gel, acamprosate, and
lovastatin) are undergoing active evaluation. Should
any of these candidates be approved in the future,
early diagnosis would prove even more beneficial.
Strengths and limitations
To our knowledge, this is the largest cohort of
Chinese FXS patients reported to date. Because most
FXS testing was performed at our centre, potential
disease prevalence can be inferred. Our study offers
a longitudinal perspective regarding the disease
course and highlights areas for improvement in
health supervision and management. Furthermore,
we examined the landscape of cascade screening and
prenatal diagnosis in our specific cultural setting.
However, this was a retrospective study and thus
largely dependent on clinician-reported findings.
The diagnostic yield may have been influenced by the
secular trend of an increasing number of referrals for
developmental delay. Furthermore, it was difficult
to implement standardised diagnostic instruments
for certain co-morbidities. Some patients had
inadequate information or were lost to follow-up in
the public sector. Finally, the lack of a standardised
surveillance protocol for FXS contributed to
potential confirmation bias.
Conclusion
In our study, we explored the diagnostic yield of
FXS testing, as well as cascade testing and prenatal
diagnosis in families with FXS in Hong Kong. Our
study provides insights into the clinical features and
co-morbidities of FXS in the largest cohort of Chinese
patients reported to date. There has been improved
awareness of children’s developmental needs, as
demonstrated by a trend towards earlier diagnosis,
but no local surveillance protocols exist for patients
with FXS. The high prevalences of neurobehavioural
and medical co-morbidities highlight the need for prompt diagnosis and structured health
management. We recommend increased awareness
of the multisystemic approach and targeted
treatments currently under investigation, and
we propose establishing rare disease registries to
facilitate this process.
Considering the clinical utility of FXS testing
in clinical and reproductive management, we believe
it should continue to be included in the evaluation
of patients with developmental delay or ID; its role
in the diagnostic pathway should be determined by
local resources.
Author contributions
Concept or design: CWM Au, HM Luk, IFM Lo.
Acquisition of data: CWM Au, S Ho.
Analysis or interpretation of data: CWM Au.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: CWM Au, S Ho.
Analysis or interpretation of data: CWM Au.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Acknowledgement
The authors thank the patients and their families for contributing the clinical data used in this study.
Funding/support
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
This research was approved by the Central Institutional
Review Board of Hospital Authority, Hong Kong (Ref No.: PAED-2023-061). A waiver of informed patient consent was
obtained from the Board due to the retrospective nature of
the research.
References
1. Hersh JH, Saul RA; Committee on Genetics. Health
supervision for children with fragile X syndrome. Pediatrics
2011;127:994-1006. Crossref
2. Jin X, Chen L. Fragile X syndrome as a rare disease
in China—therapeutic challenges and opportunities.
Intractable Rare Dis Res 2015;4:39-48. Crossref
3. Kidd SA, Lachiewicz A, Barbouth D, et al. Fragile X
syndrome: a review of associated medical problems.
Pediatrics 2014;134:995-1005. Crossref
4. Lubala TK, Lumaka A, Kanteng G, et al. Fragile X checklists:
a meta-analysis and development of a simplified universal
clinical checklist. Mol Genet Genomic Med 2018;6:526-32. Crossref
5. Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A
rapid polymerase chain reaction–based screening method
for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol
Diagn 2008;10:43-9. Crossref
6. Coffee B, Keith K, Albizua I, et al. Incidence of fragile X
syndrome by newborn screening for methylated FMR1
DNA. Am J Hum Genet 2009;85:503-14. Crossref
7. Lévesque S, Dombrowski C, Morel ML, et al. Screening and
instability of FMR1 alleles in a prospective sample of 24,449
mother–newborn pairs from the general population. Clin
Genet 2009;76:511-23. Crossref
8. Monaghan KG, Lyon E, Spector EB. ACMG Standards and
Guidelines for fragile X testing: a revision to the disease-specific
supplements to the Standards and Guidelines for
Clinical Genetics Laboratories of the American College of
Medical Genetics and Genomics. Genet Med 2013;15:575-86. Crossref
9. Peprah E. Fragile X syndrome: the FMR1 CGG repeat
distribution among world populations. Ann Hum Genet
2012;76:178-91. Crossref
10. Spector E, Behlmann A, Kronquist K, et al. Laboratory
testing for fragile X, 2021 revision: a technical standard of
the American College of Medical Genetics and Genomics
(ACMG). Genet Med 2021;23:799-812. Crossref
11. Coffee B, Zhang F, Warren ST, Reines D. Acetylated
histones are associated with FMR1 in normal but not
fragile X–syndrome cells. Nat Genet 1999;22:98-101. Crossref
12. Crawford DC, Acuña JM, Sherman SL. FMR1 and the
fragile X syndrome: human genome epidemiology review.
Genet Med 2001;3:359-71. Crossref
13. Chiurazzi P, Pomponi MG, Willemsen R, Oostra BA,
Neri G. In vitro reactivation of the FMR1 gene involved in
fragile X syndrome. Hum Mol Genet 1998;7:109-13. Crossref
14. Sutcliffe JS, Nelson DL, Zhang F, et al. DNA methylation
represses FMR-1 transcription in fragile X syndrome. Hum
Mol Genet 1992;1:397-400. Crossref
15. Nolin SL, Brown WT, Glicksman A, et al. Expansion of
the fragile X CGG repeat in females with premutation or
intermediate alleles. Am J Hum Genet 2003;72:454-64. Crossref
16. de Vries BB, Halley DJ, Oostra BA, Niermeijer MF. The
fragile X syndrome. J Med Genet 1998;35:579-89. Crossref
17. Finucane B, Abrams L, Cronister A, Archibald AD,
Bennett RL, McConkie-Rosell A. Genetic counseling and
testing for FMR1 gene mutations: practice guidelines of
the National Society of Genetic Counselors. J Genet Couns
2012;21:752-60. Crossref
18. Bailey DB Jr, Raspa M, Bishop E, Holiday D. No change in
the age of diagnosis for fragile X syndrome: findings from a
national parent survey. Pediatrics 2009;124:527-33. Crossref
19. Carmichael B, Pembrey M, Turner G, Barnicoat A.
Diagnosis of fragile-X syndrome: the experiences of
parents. J Intellect Disabil Res 1999;43:47-53. Crossref
20. Kraan CM, Godler DE, Amor DJ. Epigenetics of fragile X
syndrome and fragile X–related disorders. Dev Med Child
Neurol 2019;61:121-7. Crossref
21. Baker EK, Arpone M, Vera SA, et al. Intellectual functioning
and behavioural features associated with mosaicism in
fragile X syndrome. J Neurodev Disord 2019;11:41. Crossref
22. Meng L, Kaufmann WE, Frye RE, et al. The association
between mosaicism type and cognitive and behavioral
functioning among males with fragile X syndrome. Am J
Med Genet A 2022;188:858-66. Crossref
23. Nolin SL, Glicksman A, Houck GE Jr, Brown WT, Dobkin CS. Mosaicism in fragile X affected males. Am J Med Genet 1994;51:509-12. Crossref
24. Liu Y, Chen Y, Zhou X, et al. Mutational analysis of FMR1
gene in autistic children of Han ethnic [in Chinese]. Chin J
Child Health Care 2011;19:701-3.
25. Zhang X, Zhong J, Huo X, et al. Screening and genetic
diagnosis of fragile X syndrome among children with
mental retardation of unknown cause [in Chinese]. Chin J
Birth Health Hered 2010;18:38-9.
26. Centre for Health Protection, Department of Health, Hong
Kong SAR Government. Overweight and obesity. 2021.
Available from: https://www.chp.gov.hk/en/statistics/data/10/757/5513.html. Accessed 14 Jan 2024.
27. Raspa M, Bailey DB, Bishop E, Holiday D, Olmsted
M. Obesity, food selectivity, and physical activity in
individuals with fragile X syndrome. Am J Intellect Dev
Disabil 2010;115:482-95. Crossref
28. Kidd SA, Berry-Kravis E, Choo TH, et al. Improving
the diagnosis of autism spectrum disorder in fragile
X syndrome by adapting the Social Communication
Questionnaire and the Social Responsiveness Scale–2. J
Autism Dev Disord 2020;50:3276-95. Crossref
29. Ciaccio C, Fontana L, Milani D, Tabano S, Miozzo M,
Esposito S. Fragile X syndrome: a review of clinical and
molecular diagnoses. Ital J Pediatr 2017;43:39. Crossref
30. Berry-Kravis E, Filipink RA, Frye RE, et al. Seizures in
fragile X syndrome: associations and longitudinal analysis
of a large clinic-based cohort. Front Pediatr 2021;9:736255. Crossref
31. Loehr JP, Synhorst DP, Wolfe RR, Hagerman RJ. Aortic
root dilatation and mitral valve prolapse in the fragile X
syndrome. Am J Med Genet 1986;23:189-94. Crossref
32. Mei L, Hu C, Li D, et al. The incidence and clinical
characteristics of fragile X syndrome in China. Front
Pediatr 2023;11:1064104. Crossref
33. Charalsawadi C, Wirojanan J, Jaruratanasirikul S,
Ruangdaraganon N, Geater A, Limprasert P. Common
clinical characteristics and rare medical problems of fragile
X syndrome in Thai patients and review of the literature.
Int J Pediatr 2017;2017:9318346. Crossref
34. Pang CP, Poon PM, Chen QL, et al. Trinucleotide CGG
repeat in the FMR1 gene in Chinese mentally retarded
patients. Am J Med Genet 1999;84:179-83. Crossref
35. Chen X, Wang J, Xie H, et al. Fragile X syndrome
screening in Chinese children with unknown intellectual
developmental disorder. BMC Pediatr 2015;15:77. Crossref
36. Zhong N, Ju W, Xu W, et al. Frequency of the fragile X
syndrome in Chinese mentally retarded populations is
similar to that in Caucasians. Am J Med Genet 1999;84:191-4. Crossref
37. Mullegama SV, Klein SD, Nguyen DC, et al. Is it time to
retire fragile X testing as a first-tier test for developmental
delay, intellectual disability, and autism spectrum disorder?
Genet Med 2017;19:1380-1. Crossref
38. Cheng YK, Lin CS, Kwok YK, et al. Identification of fragile X
pre-mutation carriers in the Chinese obstetric population
using a robust FMR1 polymerase chain reaction assay:
implications for screening and prenatal diagnosis. Hong
Kong Med J 2017;23:110-6. Crossref
39. Huang W, Xia Q, Luo S, et al. Distribution of fragile X
mental retardation 1 CGG repeat and flanking haplotypes
in a large Chinese population. Mol Genet Genomic Med
2015;3:172-81. Crossref
40. Genereux DP, Laird CD. Why do fragile X carrier
frequencies differ between Asian and non-Asian
populations? Genes Genet Syst 2013;88:211-24. Crossref
41. The American College of Obstetricians and Gynecologists.
Carrier Screening for Genetic Conditions. Committee
Opinion. Number 691. March 2017. Available from: https://www.acog.org/clinical/clinical-guidance/committee-opinion/articles/2017/03/carrier-screening-for-genetic-conditions. Accessed 25 Apr 2024.
42. Gregg AR, Aarabi M, Klugman S, et al. Screening for
autosomal recessive and X-linked conditions during
pregnancy and preconception: a practice resource of the
American College of Medical Genetics and Genomics
(ACMG). Genet Med 2021;23:1793-806. Crossref