Hong Kong Med J 2026;32:Epub 9 Apr 2026
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Creamy clues to monogenic hypertriglyceridaemia
Antony Fu, MB, ChB, FHKAM (Paediatrics); Cindy Chan, MB, BS, FHKAM (Paediatrics)
Department of Paediatrics and Adolescent Medicine, Princess Margaret Hospital, Hong Kong SAR, China
Corresponding author: Dr Antony Fu (antony.fu@ha.org.hk)
 Full paper in PDF
Full paper in PDF
A previously healthy 21-month-old Pakistani girl
presented to our institution in June 2025 with
a 1-year history of a pruritic papulovesicular
rash affecting her limbs and groin. Apart from
intermittent viral illnesses, there were no reported
episodes of abdominal pain, vomiting or other
symptoms suggestive of acute pancreatitis. She had
been thriving well with normal intake, urine output
and bowel habit. She was born at term by vaginal
delivery with an uneventful perinatal history. Her
parents were healthy second cousins.
Physical examination of the patient revealed
multiple discrete yellow papules over all four limbs
and the groin, sparing the face, trunk and back.
Otherwise, she appeared well with age-appropriate
growth parameters.
Two months prior to the current presentation,
the patient had been admitted with an upper
respiratory tract infection. Laboratory investigations
showed mild microcytic anaemia, but routine
biochemistry was unremarkable. The chemical pathology report noted serum turbidity requiring
clearance by high-speed centrifugation prior to
non-lipid analysis; however, this critical finding was
overlooked, and the patient was discharged home.
At the latest clinic visit, the skin lesions were
recognised as eruptive xanthomas, appearing as
small yellow papules with a creamy centre (Fig 1).
A grossly lipaemic blood sample showed a thick
creamy layer (Fig 2), raising clinical suspicion of
severe hypertriglyceridaemia. Laboratory testing
confirmed this, with triglyceride level measuring
100.6 mmol/L.
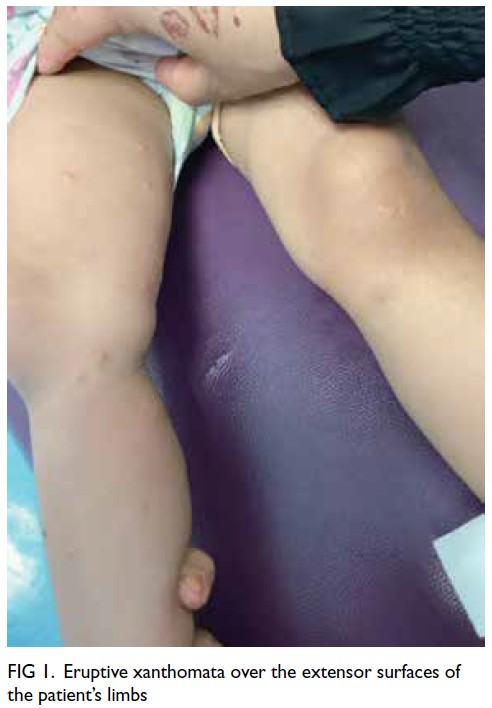
Figure 1. Eruptive xanthomata over the extensor surfaces of the patient’s limbs
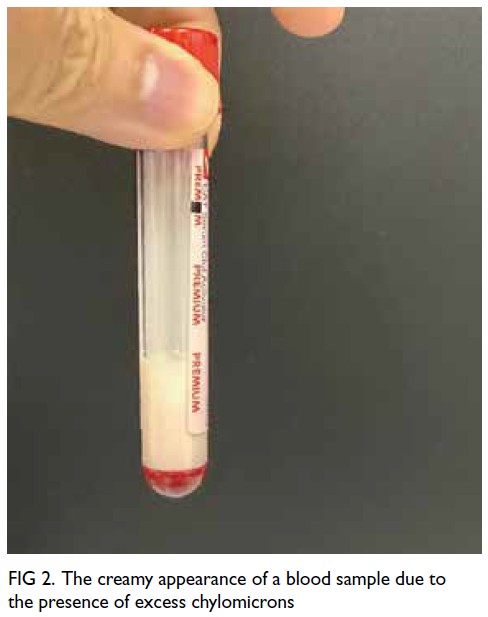
Figure 2. The creamy appearance of a blood sample due to the presence of excess chylomicrons
The patient was admitted to the paediatric
intensive care unit, kept nil by mouth and
commenced on intravenous Actrapid (Bagsværd,
Denmark) 0.1 unit/kg/hr, with a dextrose infusion to
maintain euglycaemia. After 43.5 hours of infusion
therapy, triglyceride level had reduced to 1.3 mmol/L
(Fig 3). Following intravenous insulin therapy, she
was managed with dietary restriction and essential
fatty acid supplementation. Throughout the course,
serial amylase and lipase levels remained normal.
Computed tomography excluded acute pancreatitis.
A family history revealed a nephew with monogenic hypertriglyceridaemia due to a GPIHBP1 mutation,
which was subsequently confirmed in our patient.
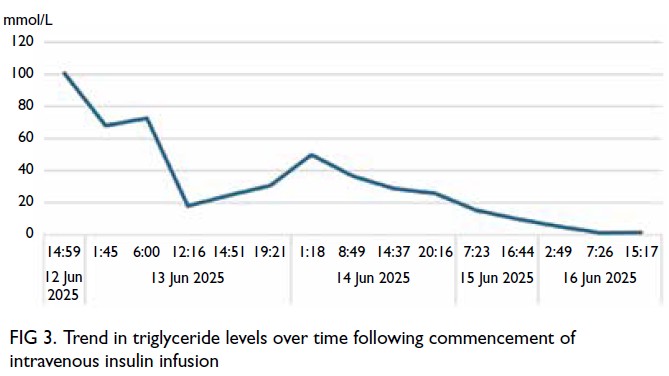
Figure 3. Trend in triglyceride levels over time following commencement of intravenous insulin infusion
Monogenic hypertriglyceridaemia, also known
as type I hyperlipidaemia, is an uncommon autosomal
recessive condition most often resulting from
LPL gene mutations that impair lipoprotein lipase
function.1 The disorder usually manifests in childhood
with recurrent abdominal pain, pancreatitis, or
characteristic features such as eruptive xanthomas,
lipaemia retinalis, and hepatosplenomegaly. Without
appropriate management, recurrent pancreatitis
may progress to chronic disease with subsequent
exocrine and endocrine insufficiency. Clinical
manifestations typically emerge before 10 years of
age, and approximately one quarter of cases present
within the first year of life.2
Acute pancreatitis typically arises when serum
triglyceride concentrations exceed 11.3 mmol/L.3 4
Management involves fasting, intravenous fluid
support, and continuous insulin infusion. This
activates lipoprotein lipase, accelerating triglyceride
clearance—reducing levels within 24 hours by
approximately 40% with insulin alone and up to 80%
when combined with fasting.5 Despite this, some
individuals, as in our case, remain asymptomatic even
with extreme hypertriglyceridaemia.6 In retrospect,
earlier recognition of these ‘creamy’ clues—eruptive
xanthomas and lipaemic serum—could have enabled
a timelier diagnosis of this rare lipid disorder.
Author contributions
Concept or design: A Fu.
Acquisition of data: Both authors.
Analysis or interpretation of data: Both authors.
Drafting of the manuscript: A Fu.
Critical revision of the manuscript for important intellectual content: A Fu.
Acquisition of data: Both authors.
Analysis or interpretation of data: Both authors.
Drafting of the manuscript: A Fu.
Critical revision of the manuscript for important intellectual content: A Fu.
Both authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
Both authors have disclosed no conflicts of interest.
Acknowledgement
The authors thank Dr Doris Ching and her team from the
Department of Chemical Pathology, Princess Margaret
Hospital for their unwavering support in facilitating the
molecular diagnosis of the patient.
Funding/support
This study received no specific grant from any funding agency
in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of
Helsinki. The parents of the patient provided written consent
for all treatments and procedures, and verbal consent for
publication, including the publication of the accompanying
clinical images.
References
1. Chyzhyk V, Brown AS. Familial chylomicronemia syndrome: a rare but devastating autosomal recessive disorder characterized by refractory hypertriglyceridemia and recurrent pancreatitis. Trends Cardiovasc Med 2020;30:80-5.
Crossref
2. Mustafa M, Almheiri M. Six-year follow-up of a child with familial chylomicronemia syndrome: disease course and effectiveness of gemfibrozil treatment–case report and literature review. Ann Pediatr Endocrinol Metab 2024;29:130-4.
Crossref
3. Krishnamurthy A, Homan E, Kim SM. Diagnosis, evaluation, and management of severe hypertriglyceridemia. Curr Cardiovasc Risk Rep 2025;19:6.
Crossref
4. Valaiyapathi B, Ashraf AP. Hospital management of severe hypertriglyceridemia in children. Curr Pediatr Rev 2017;13:225-31.
Crossref
5. Witztum JL, Gaudet D, Freedman SD, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N Engl J Med 2019;381:531-42.
Crossref
6. Schaefer EW, Leung A, Kravarusic J, Stone NJ. Management of severe hypertriglyceridemia in the hospital: a review. J Hosp Med 2012;7:431-8.
Crossref