Hong Kong Med J 2026;32:Epub 28 Jan 2026
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Early prenatal detection of autosomal dominant
skeletal dysplasia using first-trimester ultrasound and cell-free fetal DNA screening: three case reports
Ye Cao, PhD, FACMG1,2; Yvonne KY Cheng, MSc (Medical Genetics), FHKAM (Obstetrics and Gynaecology)1; TY Leung, MD, FHKAM (Obstetrics and Gynaecology)1,2; Shuwen Xue, MPhil, PhD1,2; Yuting Zheng, MPhil1,2; KW Choy, MSc (Med), PhD1,2; Winnie CW Chu, MD, FHKAM (Radiology)3; HM Luk, MD, FHKAM (Paediatrics)4; KM Law, FRCOG, FHKAM (Obstetrics and Gynaecology)1; YH Ting, FRCOG, FHKAM (Obstetrics and Gynaecology)1
1 Department of Obstetrics and Gynaecology, The Chinese University of Hong Kong, Hong Kong SAR, China
2 Shenzhen Research Institute, The Chinese University of Hong Kong, Shenzhen, China
3 Department of Imaging and Interventional Radiology, The Chinese University of Hong Kong, Hong Kong SAR, China
4 Department of Clinical Genetics, Hong Kong Children’s Hospital, Hong Kong SAR, China
Corresponding author: Dr YH Ting (tingyh@cuhk.edu.hk)
 Full paper in PDF
Full paper in PDF
Case presentations
Case 1 (Family 1)
A primigravida attended our fetal medicine clinic
(FMC) in March 2015 at 12 weeks’ gestation for first-trimester
(T1) Down syndrome screening. Ultrasound
examination revealed an absent nasal bone (NB). A
morphology scan at 20 weeks confirmed this finding,
along with bilateral non-ossified parietal bones, 11
pairs of ribs, and shortened femur and humerus.
Amniocentesis revealed a normal chromosomal
microarray. The couple opted for termination of
pregnancy at 22 weeks. A computed tomography
babygram confirmed the ultrasound findings and
also showed bilaterally absent clavicles, hinting at a
diagnosis of cleidocranial dysplasia (CCD). Targeted
sequencing of the RUNX2 gene on the amniotic fluid
sample revealed a de novo heterozygous pathogenic
missense variant, c.674G>A (p.Arg225Gln),
confirming the diagnosis. Multimodal prenatal and
genetic findings were illustrated in Figure 1.
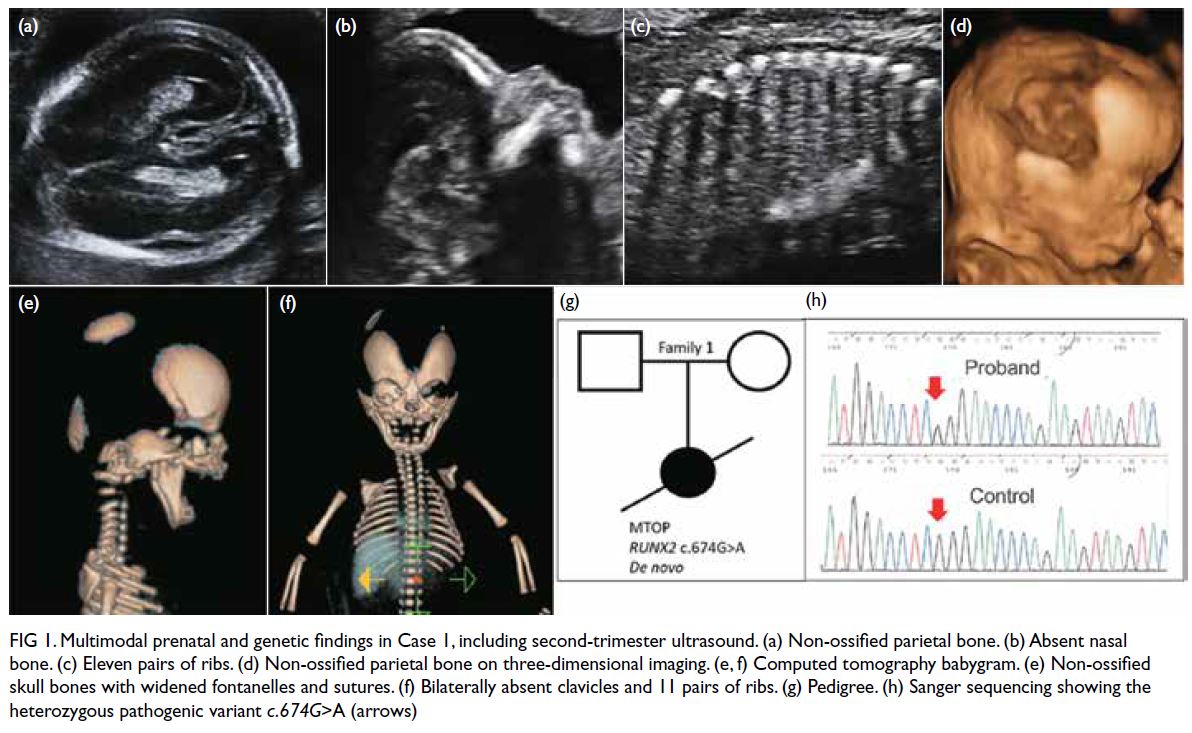
Figure 1. Multimodal prenatal and genetic findings in Case 1, including second-trimester ultrasound. (a) Non-ossified parietal bone. (b) Absent nasal bone. (c) Eleven pairs of ribs. (d) Non-ossified parietal bone on three-dimensional imaging. (e, f) Computed tomography babygram. (e) Non-ossified skull bones with widened fontanelles and sutures. (f) Bilaterally absent clavicles and 11 pairs of ribs. (g) Pedigree. (h) Sanger sequencing showing the heterozygous pathogenic variant c.674G>A (arrows)
Cases 2 and 3 (Family 2)
A primigravida attended the FMC in July 2022
at 12 weeks’ gestation for non-invasive prenatal
screening (NIPS) for fetal aneuploidy. Ultrasound
showed non-ossified skull bones (SB) and reduced
spine ossification, but both clavicles were present. A
review of the paternal history revealed that he had
features of CCD, including the ability to approximate
his shoulders, similar to a character in an American
drama with diagnosed CCD. Molecular testing for
CCD showed a pathogenic nonsense variant in the
RUNX2 gene, c.577C>T (p.Arg193Ter), confirming
the diagnosis. The fetus was thus suspected to have the
same genetic problem. The couple declined invasive
genetic testing. Instead, NIPS was performed using a
novel technique known as coordinative allele-aware target enrichment sequencing (COATE-seq). This
facilitated concomitant screening for chromosomal
and monogenic disorders, encompassing 10
aneuploidies, 12 microdeletions and 64 monogenic
disorders including RUNX2-related diseases (online supplementary Table 1). Results showed that the
fetus was at high risk of having a pathogenic variant
in the RUNX2 gene c.577C>T (p.Arg193Ter). Serial
ultrasound showed normal SB ossification but
with widened sutures, normal spine ossification,
and mildly shortened clavicles with a normal S
shape. A male infant was delivered at 39 weeks.
Skeletal survey showed a persistent metopic suture,
widened anterior fontanelle, 11 pairs of ribs, delayed
ossification of pubic bones with widely spaced public
symphysis, but both clavicles were present. Targeted
RUNX2 variant analysis on the cord blood sample
validated the presence of the paternal heterozygous
pathogenic variant.
In the same patient’s second pregnancy, she
attended the FMC at 12 weeks in January 2024 where
ultrasound showed hypoplastic clavicles, non-ossified
SBs and reduced spine ossification. The NIPS
using COATE-seq showed that the fetus was at high
risk of having the same pathogenic RUNX2 variant,
c.577C>T (p.Arg193Ter). The couple declined
invasive confirmatory testing. Serial ultrasound
showed non-ossified SBs with widened sutures
and fontanelle, a thin NB, very short clavicles with
loss of normal S shape, 11 pairs of ribs, and mildly
shortened long bones. A female infant was delivered
at 38 weeks. Skeletal survey revealed bilateral
hypoplastic clavicles and 11 pairs of ribs. Targeted
RUNX2 variant analysis of the cord blood sample
validated the presence of the paternal heterozygous
pathogenic variant, confirming the diagnosis.
Imaging and genetic findings are illustrated in Figure 2.
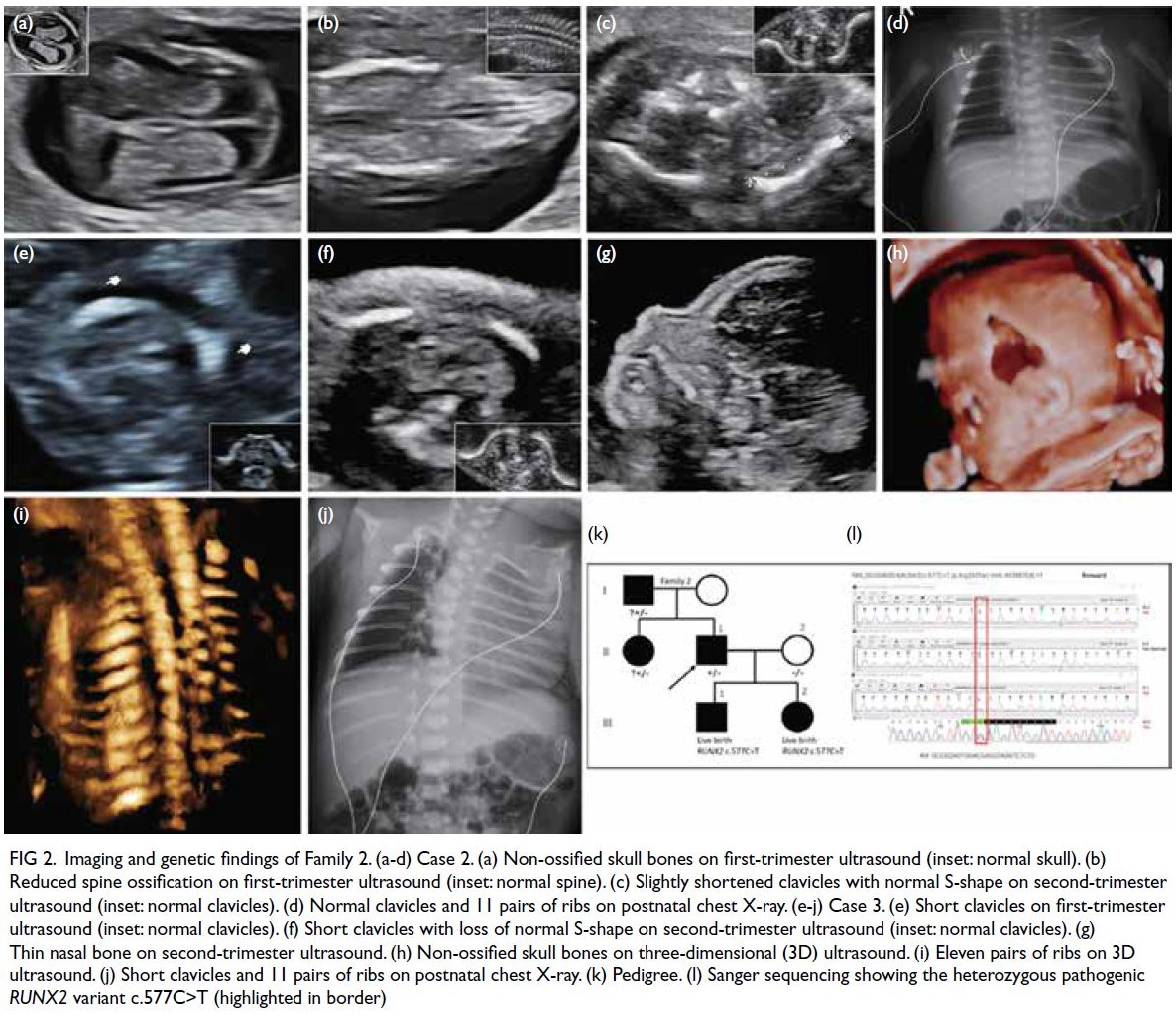
Figure 2. Imaging and genetic findings of Family 2. (a-d) Case 2. (a) Non-ossified skull bones on first-trimester ultrasound (inset: normal skull). (b) Reduced spine ossification on first-trimester ultrasound (inset: normal spine). (c) Slightly shortened clavicles with normal S-shape on second-trimester ultrasound (inset: normal clavicles). (d) Normal clavicles and 11 pairs of ribs on postnatal chest X-ray. (e-j) Case 3. (e) Short clavicles on first-trimester ultrasound (inset: normal clavicles). (f) Short clavicles with loss of normal S-shape on second-trimester ultrasound (inset: normal clavicles). (g) Thin nasal bone on second-trimester ultrasound. (h) Non-ossified skull bones on three-dimensional (3D) ultrasound. (i) Eleven pairs of ribs on 3D ultrasound. (j) Short clavicles and 11 pairs of ribs on postnatal chest X-ray. (k) Pedigree. (l) Sanger sequencing showing the heterozygous pathogenic RUNX2 variant c.577C>T (highlighted in border)
Discussion
Cleidocranial dysplasia is a rare autosomal dominant
skeletal dysplasia characterised by the classic
triad of absent or hypoplastic clavicles, delayed
ossification of the cranial bones with delayed
closure of sutures and fontanelles, and dental
abnormalities.1 Approximately two-thirds of cases
are caused by RUNX2 gene mutations, with the
remaining one-third resulting from copy number
variations, translocations, or inversions involving
the RUNX2 locus.2 The RUNX2 gene, located
on chromosome 6p21, encodes a transcription
factor that regulates osteoblast differentiation and
chondrocyte maturation.3 Haploinsufficiency of
RUNX2 gene leads to delayed intramembranous and
endochondral ossification.3 The skull and clavicles,
formed by intramembranous ossification, are
therefore the most frequently affected.3
Prenatal diagnosis of CCD is rare. Including
our three cases, only 22 cases have been reported
to date (online supplementary Table 2). Most had
affected family members, hinting at the diagnosis.
Most were diagnosed based on clinical findings, with
only 10 cases having a molecular diagnosis of RUNX2
gene defects. This highlights the pivotal role of
prenatal ultrasound in identifying the characteristic features, namely, absent or hypoplastic clavicles,
absent or inadequate SB ossification with wide
fontanelles and sutures, and shortened long bones
and absent NB. Among these, clavicular defect is
the most characteristic. All three cases in our series
had these typical features, detected during the first
trimester, with an additional novel finding of 11
pairs of ribs. Nevertheless, the prenatal detection of
CCD can be difficult as ultrasound features may be
subtle. Although clavicles can be visualised during
T1 ultrasound, they are not routinely examined.
Conversely, absent NB, a marker for aneuploidy and
routinely assessed during T1 nuchal translucency
measurement, may be an important clue that
prompts further examination of the clavicles and
skull. With a positive family history, prenatal
detection of inherited CCD by ultrasound may be
more feasible. However, this can remain challenging
as pathogenic RUNX2 variants exhibit complete
penetrance but variable expressivity.1 Within the
same family, one affected fetus may present with a
subtle phenotype while another may show more
pronounced manifestations, as illustrated by the
two siblings in Family 2. Therefore, meticulous
ultrasound is imperative in pregnancies at risk of
CCD.
When CCD is suspected, invasive genetic testing
is usually recommended to confirm the diagnosis
through targeted RUNX2 variant analysis. However,
invasive testing is associated with 0.1% to 0.2% risk of
procedure-related fetal loss.4 As CCD rarely results
in severe disability, many parents, particularly
affected ones, may not consider termination of
pregnancy and may choose to avoid invasive testing.
In such cases, the new NIPS approach, COATE-seq,
provides a viable diagnostic alternative.5 Its
performance in high-risk pregnancies has been
validated, demonstrating 98.5% sensitivity and
99.3% specificity compared with standard diagnostic
methods.6 The two cases in Family 2 represent the
first report of prenatal detection of CCD through the
identification of a pathogenic RUNX2 variant using
this novel technique. These cases highlight the great potential of combining T1 ultrasound with NIPS for
early, non-invasive prenatal detection. This powerful
non-invasive approach may also be applicable to
other autosomal dominant skeletal dysplasia and
monogenic disorders.
Author contributions
Concept or design: KW Choy, YH Ting.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: Y Cao, YH Ting.
Critical revision of the manuscript for important intellectual content: Y Cao, YH Ting.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: Y Cao, YH Ting.
Critical revision of the manuscript for important intellectual content: Y Cao, YH Ting.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Acknowledgement
The authors thank the affected families for participating in
and supporting this study.
Funding/support
This study was supported by the National Key Research
and Development Program of China (Grant No.:
2023YFC2705603). The funder had no role in study design,
data collection/analysis/interpretation or manuscript
preparation.
Ethics approval
This study was approved by the Joint Chinese University of
Hong Kong–New Territories East Cluster Clinical Research
Ethics Committee, Hong Kong (Ref No.: 2017.442). Written
informed consent was obtained from the families for
publication of clinical details and images.
Supplementary material
The supplementary material was provided by the authors and
some information may not have been peer reviewed. Accepted
supplementary material will be published as submitted by the
authors, without any editing or formatting. Any opinions
or recommendations discussed are solely those of the
author(s) and are not endorsed by the Hong Kong Academy
of Medicine and the Hong Kong Medical Association. The Hong Kong Academy of Medicine and the Hong Kong
Medical Association disclaim all liability and responsibility
arising from any reliance placed on the content.
References
1. Machol K, Mendoza-Londono R, Lee B. Cleidocranial
dysplasia spectrum disorder. 3 Jan 2006 [updated 13 Apr
2023]. In: Adam MP, Feldman J, Mirzaa GM, editors.
GeneReviews. Seattle (WA): University of Washington;
1993.
2. Motaei J, Salmaninejad A, Jamali E, et al. Molecular
genetics of cleidocranial dysplasia. Fetal Pediatr Pathol
2021;40:442-54. Crossref
3. Hassan NM, Dhillon A, Huang B. Cleidocranial dysplasia:
clinical overview and genetic considerations. Pediatr Dent
J 2016;26:45-50. Crossref
4. Akolekar R, Beta J, Picciarelli G, Ogilvie C, D’Antonio F.
Procedure-related risk of miscarriage following
amniocentesis and chorionic villus sampling: a systematic
review and meta-analysis. Ultrasound Obstet Gynecol
2015;45:16-26. Crossref
5. Xu C, Li J, Chen S, et al. Genetic deconvolution of fetal and
maternal cell-free DNA in maternal plasma enables next-generation
non-invasive prenatal screening. Cell Discov
2022;8:109. Crossref
6. Zhang J, Wu Y, Chen S, et al. Prospective prenatal cell-free
DNA screening for genetic conditions of heterogenous
etiologies. Nat Med 2024;30:470-9. Crossref