Hong Kong Med J 2026;32:Epub 20 Jan 2026
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Pneumonia-associated inflammatory myofibroblastic tumour: a case report
Xiuxin Mo1; Yuchun Zhuang1; Liming Zhang1; Chengcheng Chen2
1 Department of Thoracic Cardiovascular Surgery, Weifang Second People’s Hospital, Weifang, China
2 Department of Radiology, People’s Hospital of Rizhao, Rizhao, China
Corresponding author: Dr Chengcheng Chen (chengcheng9987@163.com)
 Full paper in PDF
Full paper in PDF
Case presentation
A 42-year-old woman was admitted to Weifang
Second People’s Hospital on 20 June 2024 following
an incidental finding of a pulmonary nodule (21 × 27 mm2) during a routine physical examination 1
year previously. Although serial imaging over the
following year showed stable size and morphology,
suggesting a benign nature, malignancy remained
possible. The patient had no significant medical,
family, or psychosocial history, and denied tobacco
and alcohol use. Preoperative evaluation included
fine-needle aspiration cytology, which revealed
spindle cells with lymphoplasmacytic infiltration.
Contrast-enhanced chest computed tomography
demonstrated a lobulated left upper lobe nodule
with heterogeneous enhancement and partial
bronchial obstruction (Fig 1). Magnetic resonance
imaging of the brain and abdominal ultrasound
showed no metastasis. Based on the above
investigations and considering the patient’s financial
circumstances, a positron emission tomography
scan was not performed. Tumour marker levels
were within the normal range—neuron-specific
enolase: 13.06 ng/mL, carbohydrate antigen 19-9:
9.76 U/mL, carcinoembryonic antigen: 1.54 ng/mL,
cytokeratin 19 fragment: 1.23 ng/mL, and squamous
cell carcinoma antigen: 0.51 ng/mL. Thoracoscopic
left upper lobectomy was performed on 22 June
2024. Histopathology revealed proliferating spindle
myofibroblasts/fibroblasts with lymphoplasmacytic
infiltration and focal mucin deposition (Fig 2).
Immunohistochemistry confirmed inflammatory
myofibroblastic tumour (IMT): positive for
cytokeratin, vimentin, smooth muscle actin (SMA),
and epithelial membrane antigen; STAT6 (signal
transducer and activator of transcription 6) negative
with a Ki-67 index of 30%. The patient recovered
well, with no recurrence at 3-month follow-up,
although long-term surveillance was recommended.
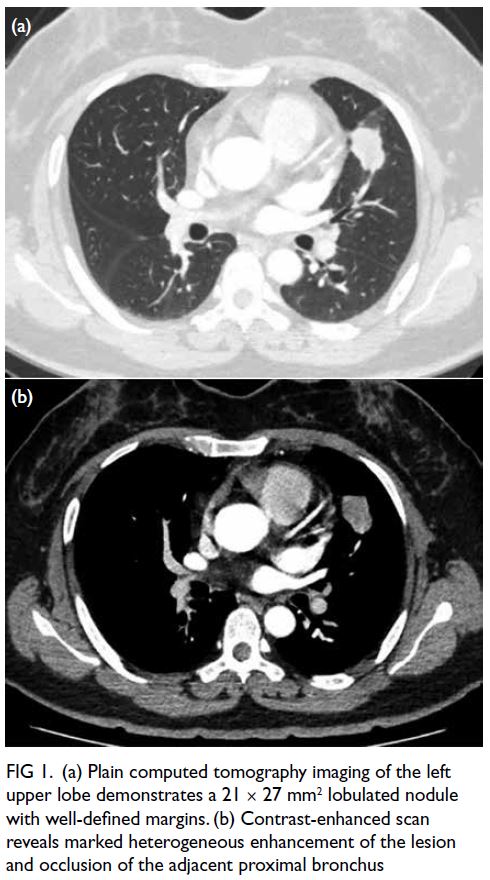
Figure 1. (a) Plain computed tomography imaging of the left upper lobe demonstrates a 21 × 27 mm2 lobulated nodule with well-defined margins. (b) Contrast-enhanced scan reveals marked heterogeneous enhancement of the lesion and occlusion of the adjacent proximal bronchus
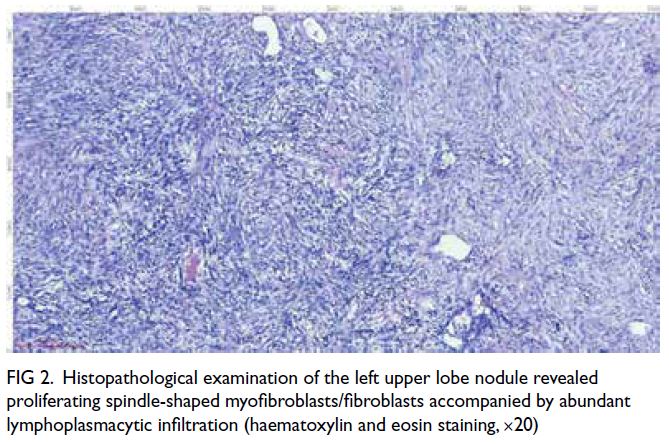
Figure 2. Histopathological examination of the left upper lobe nodule revealed proliferating spindle-shaped myofibroblasts/fibroblasts accompanied by abundant lymphoplasmacytic infiltration (haematoxylin and eosin staining, ×20)
Discussion
Inflammatory myofibroblastic tumour, originally
termed inflammatory pseudotumour (IPT) in
1939, has been reclassified through molecular
insights from a reactive proliferation to a true neoplasm.1 Although IPT remains a non-neoplastic
inflammatory lesion with regression potential, IMT
is now defined as a clonal neoplasm composed
of myofibroblastic spindle cells within a plasma
cell/lymphocyte/eosinophil-rich stroma. This
distinction is crucial clinically since IMT exhibits
local invasiveness and recurrence risk, unlike IPT’s
benign course.2
Inflammatory myofibroblastic tumour is a
rare mesenchymal neoplasm that primarily affects
children and young adults, with lower incidence
in adults.3 Its broad anatomical distribution
most commonly involves the lungs (0.7% of
pulmonary tumours)4 and the abdomen/mesentery/retroperitoneum; rare sites include the oesophagus,
cardiac chambers, and adrenal glands. As a
borderline malignancy, recurrence rates differ by
site (pulmonary 2% vs extrapulmonary 25%), with
less than 5% risk of distant metastasis.5 Symptoms
vary anatomically: pulmonary cases may present
with cough or haemoptysis (including incidental
detection), abdominal lesions may cause pain or
obstruction, while systemic symptoms include fever
and weight loss. Pulmonary IMTs, as observed in
our patient, may present with cough, atypical chest
pain, haemoptysis, or dyspnoea, although incidental
detection during routine health screening, as in our
case, is not uncommon.
The non-specific radiological features of IMT
pose significant diagnostic challenges, necessitating
histopathological confirmation. In our patient, the
nodule was identified during a routine physical
examination 1 year prior to admission, and serial
imaging demonstrated stable lesion size. This
supported a benign nature but did not entirely
exclude malignancy. Although minimally invasive
techniques such as fine-needle aspiration biopsy
and bronchoscopic sampling are often attempted,
these methods frequently yield insufficient
tissue for definitive diagnosis. Complete surgical
resection therefore remains the gold standard
for both diagnostic confirmation and therapeutic
intervention.
Histopathological examination typically reveals spindle-shaped myofibroblastic proliferation
within variable stromal matrices (myxoid,
collagenous, or calcified patterns), accompanied
by a polymorphic inflammatory infiltrate. In
our patient, the histopathological features were
consistent with IMT, showing proliferating spindle-shaped
myofibroblasts/fibroblasts with abundant
lymphoplasmacytic infiltration and focal mucin
deposition. The diagnosis was further supported by
immunohistochemical findings, including positivity
for cytokeratin, vimentin, SMA, and epithelial
membrane antigen, although anaplastic lymphoma
kinase (ALK) and STAT6 were negative.
Molecular studies have identified
chromosomal 2p23 translocations in approximately
50% of IMT cases, leading to constitutive activation
of ALK pathways.6 This genetic aberration
correlates with tumour aggressiveness and local
recurrence, supporting IMT’s classification as a
true neoplasm rather than a reactive pseudotumour.
Immunophenotypically, most IMTs express
mesenchymal markers such as ALK (cytoplasmic/membranous), caldesmon, desmin, and SMA,
with ALK reactivity aiding differentiation from
histological mimics. Notably, our case showed an
atypical immunoprofile with SMA positivity and ALK
negativity, reflecting the phenotypic heterogeneity
and the need for comprehensive molecular profiling
in challenging cases.
Therapeutic strategies for IMT depend on
disease stage and resectability. For localised lesions,
complete surgical resection (R0 margins) achieves a
2% recurrence rate, whereas incomplete resection
(R1/R2) increases recurrence risk to 60% (P<0.01).7
In our patient, thoracoscopic left upper lobectomy
was performed with negative surgical margins, and
no tumour recurrence was observed during the initial
3-month postoperative follow-up. Nonetheless,
longer-term surveillance is recommended to confirm
the absence of tumour recurrence. Non-resectable
or recurrent cases require multimodal approaches,
including radiotherapy (45-50 Gy), platinum-based
chemotherapy, and ALK inhibitors for ALK-positive
subtypes.
Emerging molecular insights have identified
ALK rearrangements as key oncogenic drivers,
positioning ALK-targeted therapies as both
diagnostic and therapeutic tools.8 Clinical trials have
demonstrated the efficacy of crizotinib: an initial
phase 1 study (NCT01121588)9 achieved a 42.9%
partial response rate in refractory paediatric/young
adult IMTs (n=7), while cohort expansion (n=14)
improved the overall response rate (ORR) to 86% (36%
complete responses). Japanese studies corroborate
these findings, with 100% ORR (1 complete response,
2 partial responses) in ALK-rearranged IMTs
treated with crizotinib or alectinib.10 Nonetheless,
therapeutic heterogeneity (ORR: 36%-100%), small sample sizes, and geographical bias necessitate
standardised multicentre trials to validate efficacy
and durability.
In summary, IMT represents a rare borderline
neoplasm with intermediate malignant potential,
distinct from the historically described IPT. The
present case highlights the importance of accurate
histopathological and immunohistochemical
diagnosis, particularly in immunophenotypically
atypical lesions, and underscores the need for long-term
follow-up to monitor for potential recurrence.
Author contributions
Concept or design: X Mo.
Acquisition of data: Y Zhuang.
Analysis or interpretation of data: L Zhang.
Drafting of the manuscript: X Mo.
Critical revision of the manuscript for important intellectual content: C Chen.
Acquisition of data: Y Zhuang.
Analysis or interpretation of data: L Zhang.
Drafting of the manuscript: X Mo.
Critical revision of the manuscript for important intellectual content: C Chen.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study was supported by the Science and Technology
Development Project of Weifang (Ref No.: 2024YX077) and
Weifang Youth Medical Talent Cultivation Support Program,
China. The funders had no role in the study design, data
collection/analysis/interpretation, or manuscript preparation.
Ethics approval
This study was approved by the Ethics Committee of Weifang
Second People’s Hospital, China (Ref No.: KY2024-077-01)
and was conducted in accordance with the Declaration of
Helsinki. The patient provided written informed consent for
participation and publication of this case report, including the
accompanying clinical images.
References
1. Höhne S, Milzsch M, Adams J, Kunze C, Finke R. Inflammatory pseudotumor (IPT) and inflammatory myofibroblastic tumor (IMT): a representative literature review occasioned by a rare IMT of the transverse colon in a 9-year-old child. Tumori 2015;101:249-56. Crossref
2. Sagar AE, Jimenez CA, Shannon VR. Clinical and histopathologic correlates and management strategies for inflammatory myofibroblastic tumor of the lung. A case series and review of the literature. Med Oncol 2018;35:102. Crossref
3. Kiratli H, Uzun S, Varan A, Akyüz C, Orhan D.
Management of anaplastic lymphoma kinase positive
orbito-conjunctival inflammatory myofibroblastic tumor
with crizotinib. J AAPOS 2016;20:260-3. Crossref
4. Surabhi VR, Chua S, Patel RP, Takahashi N, Lalwani N,
Prasad SR. Inflammatory myofibroblastic tumors: current
update. Radiol Clin North Am 2016;54:553-63. Crossref
5. Bedi D, Clark BZ, Carter GJ, et al. Prognostic significance
of three-tiered World Health Organization classification
of phyllodes tumor and correlation to Singapore General
Hospital nomogram. Am J Clin Pathol 2022;158:362-71. Crossref
6. Pacheco E, Llorente JL, López-Hernández A, et al. Absence
of chromosomal translocations and protein expression of
ALK in sinonasal adenocarcinomas [in English, Spanish].
Acta Otorrinolaringol Esp 2017;68:9-14. Crossref
7. Baldi GG, Brahmi M, Lo Vullo S, et al. The activity of
chemotherapy in inflammatory myofibroblastic tumors: a
multicenter, European retrospective case series analysis.
Oncologist 2020;25:e1777-84. Crossref
8. Mossé YP, Lim MS, Voss SD, et al. Safety and activity of
crizotinib for paediatric patients with refractory solid
tumours or anaplastic large-cell lymphoma: a Children’s
Oncology Group phase 1 consortium study. Lancet Oncol
2013;14:472-80. Crossref
9. Antoniu SA. Crizotinib for EML4-ALK positive lung
adenocarcinoma: a hope for the advanced disease?
Evaluation of Kwak EL, Bang YJ, Camidge DR, et al.
Anaplastic lymphoma kinase inhibition in non–small-cell
lung cancer. N Engl J Med 2010;363(18):1693-703. Expert
Opin Ther Targets 2011;15:351-3. Crossref
10. Takeyasu Y, Okuma HS, Kojima Y, et al. Impact of ALK
inhibitors in patients with ALK-rearranged nonlung solid
tumors. JCO Precis Oncol 2021;5:PO.20.00383. Crossref