Hong Kong Med J 2025;31:Epub 28 Aug 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Presentation, management, and clinical outcomes
of von Hippel–Lindau syndrome
Athena YH Lee, MB, ChB1,2 #; David KW Leung, MB, ChB, FRCS1 #; CH Leung, MSc1; Kelly HY Tsang1; Alvina Yiu1; Chloe YK Ho1; Jason MK Ho, FHKAM (Surgery), FRCSEd (Neurosurgery)3; CF Ng, MD, FHKAM (Surgery)1,4
1 Division of Urology, Department of Surgery, Faculty of Medicine, The Chinese University of Hong Kong, Hong Kong SAR, China
2 Cardio-Oncology Research Unit, Cardiovascular Analytics Group, Hong Kong, China–UK Collaboration, Hong Kong SAR, China
3 Division of Neurosurgery, Department of Surgery, Tuen Mun Hospital, Hong Kong SAR, China
4 SH Ho Urology Centre, The Chinese University of Hong Kong, Hong Kong SAR, China
# Equal contribution
Corresponding author: Prof CF Ng (ngcf@surgery.cuhk.edu.hk)
 Full paper in PDF
Full paper in PDF
Abstract
Introduction: von Hippel–Lindau (VHL) syndrome
is a rare autosomal dominant genetic disorder
that typically leads to the development of multiple
tumours in various organs. This study describes the
lifetime journey of VHL patients in terms of their
hospitalisation, surgery, and functional impairment,
and aims to examine the local presentation patterns,
treatment courses, and clinical outcomes associated
with the condition.
Methods: Thirty-two patients with VHL syndrome
(mean age=27.9 ± 12.6 years) were retrospectively
identified from five local public hospitals managed
between 1 January 1993 and 30 September 2024,
with a follow-up duration of 18.0 ± 10.8 years.
Patient demographics, disease presentation, length
of hospital stay, and treatments received were
recorded and analysed.
Results: Over a total of 575.9 person-years, 17
patients (53.1%) developed renal tumours and 10
(31.3%) underwent partial or radical nephrectomy.
Twenty-four patients (75.0%) underwent central
nervous system (CNS) surgery for haemangioma.
Eleven patients (34.4%) had phaeochromocytoma,
and eight (25.0%) underwent adrenalectomy. Nine
patients (28.1%) had retinal haemangioma. During
the study period, 368 emergency department visits,
1209 inpatient admissions, 192 intensive care unit
days, and 5635 hospitalisation days were recorded.
In total, 116 surgeries were performed involving the
kidneys (n=17), pancreas (n=6), adrenal glands (n=10),
and CNS (n=83). Six patients required dialysis; 4373
dialysis sessions were performed. Fifteen patients died. Among the nine who died of VHL syndrome,
eight had developed cerebral haemangioblastoma,
three had phaeochromocytoma, and four had renal
tumours.
Conclusion: Patients with VHL syndrome
often experience early-onset and recurrent
diseases affecting multiple organ systems,
leading to substantial morbidity and mortality.
A multidisciplinary approach, along with the
introduction of novel treatments, may improve
disease control and clinical outcomes.
New knowledge added by this study
- This study examined the disease journey of von Hippel–Lindau (VHL) patients in Hong Kong, providing insights into disease presentation patterns, the number of treatments and procedures required, treatment outcomes, and morbidity data.
- The study analysed the substantial healthcare costs incurred in managing VHL syndrome, highlighting the economic burden on healthcare systems due to repeated admissions, multidisciplinary care, long-term followup, surgeries, and other interventions, notably VHL syndrome–related renal cell carcinoma treatment and kidney dialysis.
- The study emphasises the potential benefits of novel treatments such as belzutifan in managing VHL syndrome among local patients, with promising results that could transform the treatment landscape for this rare genetic disorder, thus reducing disease burden and improving the quality of life of patients.
- Given the cross-specialty manifestations of VHL syndrome, the study underscores the importance of a multidisciplinary approach in its management, thereby demonstrating the value of collaborative care in improving clinical outcomes.
- The study’s findings may prompt policymakers to re-evaluate existing healthcare policies related to rare genetic disorders such as VHL syndrome, particularly in expanding access to innovative treatments by adding belzutifan to the Hospital Authority Drug Formulary.
- The study highlights the need for dedicated funding to establish local VHL syndrome registries, thereby supporting further clinical trials and large-scale research. The creation of patient support programmes may also contribute to a healthcare environment that addresses the unique challenges faced by VHL patients and fosters a holistic approach to care.
Introduction
von Hippel–Lindau (VHL) syndrome is a rare autosomal dominant genetic disorder characterised
by benign and malignant tumours, including
clear cell renal cell carcinoma (RCC), adrenal
phaeochromocytoma, pancreatic neuroendocrine
tumour, and retinal and central nervous system
haemangioblastoma (CNS-Hb).1 According to a
2017 study, its incidence is estimated to be one in
27 300 live births.2 The multi-system manifestations
of VHL typically require repeated admissions,
multidisciplinary care, and long-term follow-up,
placing a substantial socio-economic burden on healthcare systems. Recently, belzutifan, a second-generation
hypoxia-inducible factor (HIF)-2α
inhibitor, has shown promising results in a phase 2
study involving Western populations.3 However, its
applications and benefits for Asian patients remain
poorly understood.
This multi-centre retrospective cohort
study investigated VHL patients to examine local
presentation patterns, treatment courses, and
clinical and functional outcomes. The findings
aim to provide insight into the presentation and
management of VHL in Asian patients and, more
importantly, to inform resource allocation.
Methods
This study identified patients with VHL syndrome from five local public hospitals—Prince of Wales
Hospital, Alice Ho Miu Ling Nethersole Hospital,
North District Hospital, Tuen Mun Hospital, and
Pok Oi Hospital—managed between 1 January 1993 and 31 December 2023, with follow-up data collected
up to 30 September 2024. The Clinical Data Analysis
and Reporting System, a local online platform
recording clinical data from all public hospitals
in Hong Kong, was used for patient identification.
Patient demographics and clinical information
regarding disease course and treatment outcomes
were retrieved from the Clinical Management
System, an online database storing electronic
patient records for public hospitals in Hong Kong.
The following data were collected for each included
patient: demographic factors (age, sex, body mass
index, performance status, and co-morbidities);
disease characteristics (initial presentation, time
of diagnosis, lag time to diagnosis, number and
size of renal and extrarenal lesions, and response
or recurrence patterns); treatment details (number
and frequency of surgical or ablative interventions,
hospital length of stay, intensive care unit [ICU]
admissions, associated costs, and resultant
complications and disabilities); and health outcomes
(health-adjusted life years, quality of life estimates,
and economic parameters related to hospitalisations,
outpatient services, and medical and surgical care).
The study endpoints included rates of VHL-spectrum
disease (CNS-Hb, choroid plexus
papilloma, retinal haemangioma, endolymphatic
sac tumour, RCC, renal cyst, renal angiomyolipoma,
phaeochromocytoma, paraganglioma, pancreatic
cyst, pancreatic neuroendocrine tumour, pancreatic
adenocarcinoma, and liver cyst), emergency
department (ED) attendance, admissions, surgeries,
and functional outcomes (independent in activities
of daily living, wheelchair-bound, or bedbound).
According to the local public healthcare system
in Hong Kong, the mean cost per ambulatory emergency attendance and per hospitalisation day
was HK$750 (US$96.2) and HK$3440 (US$441),
respectively.4 The total cost of hospital attendance
was defined as the sum of ED and inpatient
attendance costs. Descriptive statistics, including
mean, standard deviation, median, and interquartile
range, were used to summarise the data.
Results
Demographics
Initially, 87 patients were identified. After manual
review of the medical records, 52 were excluded
due to incorrect diagnoses (three non-VHL, two
Cowden syndrome, 17 Peutz–Jeghers syndrome,
28 Sturge–Weber syndrome, one hamartoma, and
one duplicate record). Two additional patients were
excluded due to incomplete data, and one further
duplicate was removed. The incorrect diagnoses
were likely due to similarities and overlaps in the
diagnostic codes used for these conditions.
In total, 32 patients were deemed eligible for
inclusion, of whom 21 (65.6%) were male. The mean
age at first presentation was 27.9 ± 12.6 years and
the mean follow-up duration was 18.0 ± 10.8 years.
All patients developed tumours. Seventeen patients
(53.1%) had renal tumours, and 10 (31.3%) underwent
partial or radical nephrectomy. Twenty-four patients
(75.0%) underwent CNS surgery for haemangioma.
Eleven patients (34.4%) had phaeochromocytoma,
and eight (25.0%) underwent adrenalectomy. Retinal
haemangioma occurred in nine patients (28.1%).
Demographic and disease prevalence data within the
VHL syndrome spectrum are summarised in Table 1.
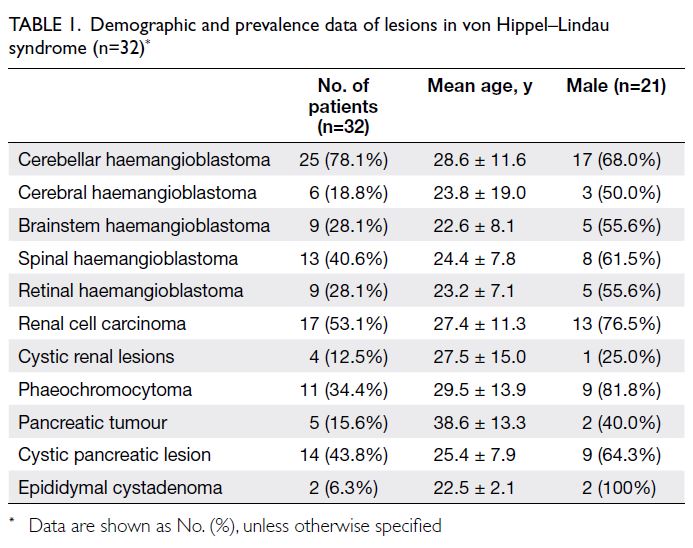
Table 1. Demographic and prevalence data of lesions in von Hippel–Lindau syndrome (n=32)
von Hippel–Lindau syndrome–related mortality
Over a total of 575.9 person-years, 15 patients died.
Causes of death were VHL syndrome in nine (60%),
pneumonia in three (20%), metastatic lung cancer
in one (6.7%), sepsis in one (6.7%), and congestive
heart failure in one (6.7%). Among those who died
of VHL syndrome–related tumours, eight had CNS
haemangioma, three had phaeochromocytoma,
and four had renal tumours. Even in patients whose
causes of death were not directly related to VHL,
strong associations were observed with the sequelae
of VHL-spectrum diseases and treatments. All
three patients who died of chest infections were
wheelchair-bound after neurosurgical treatment of
CNS-Hb; one of them required long-term steroids
following bilateral adrenalectomy. The patient who
died of sepsis had paraplegia after spinal surgery and
end-stage renal failure (ESRF) requiring peritoneal
dialysis. The source of sepsis was likely peritoneal
dialysis–related peritonitis. All-cause mortality
and VHL syndrome–related mortality over time
since presentation are shown in Figures 1 and 2, respectively.
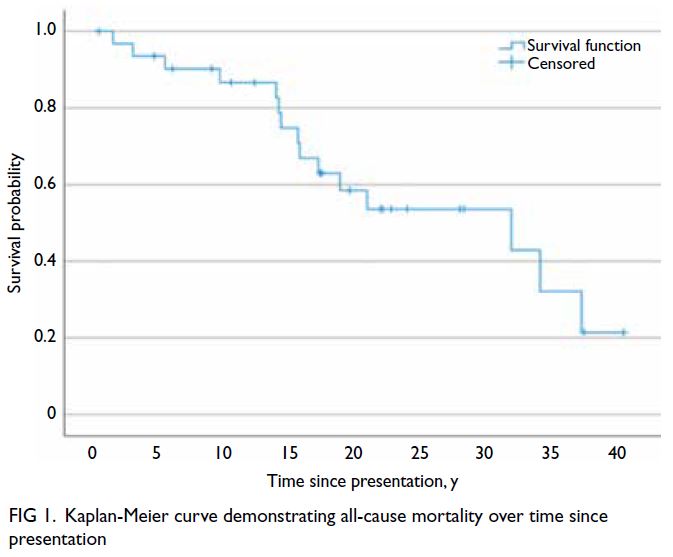
Figure 1. Kaplan-Meier curve demonstrating all-cause mortality over time since presentation
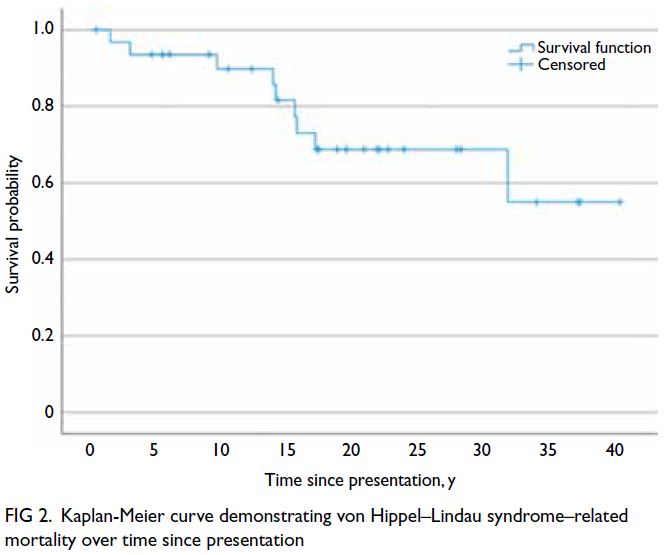
Figure 2. Kaplan-Meier curve demonstrating von Hippel–Lindau syndrome–related mortality over time since presentation
von Hippel–Lindau syndrome–related morbidity
Nine patients (28.1%) developed chronic kidney
disease, of whom six progressed to ESRF (estimated
glomerular filtration rate <15 mL/min/1.73 m2). All
six (18.8%) required renal replacement therapy—three underwent haemodialysis, one received
peritoneal dialysis, and two began peritoneal dialysis
before switching to haemodialysis.
By the last follow-up, 15 patients had died,
whereas 17 remained independent in their activities
of daily living. None of the 17 surviving patients
were wheelchair-bound or bedbound.
Belzutifan usage
Belzutifan was prescribed to three patients. The
mean age at presentation was 26.3 years, with the
youngest at 17 years and the oldest at 35 years. The
average duration from initial presentation to the
initiation of belzutifan therapy was 22.9 years. All three patients had CNS haemangioma, with one
experiencing multiple recurrences. One patient also
had phaeochromocytoma, and another had a renal
tumour. Patient characteristics are summarised in
Table 2. The duration of belzutifan therapy ranged
from 1 to 7.6 months. Of the three patients, two
required dose reductions due to adverse events—specifically, anaemia and deranged liver function.
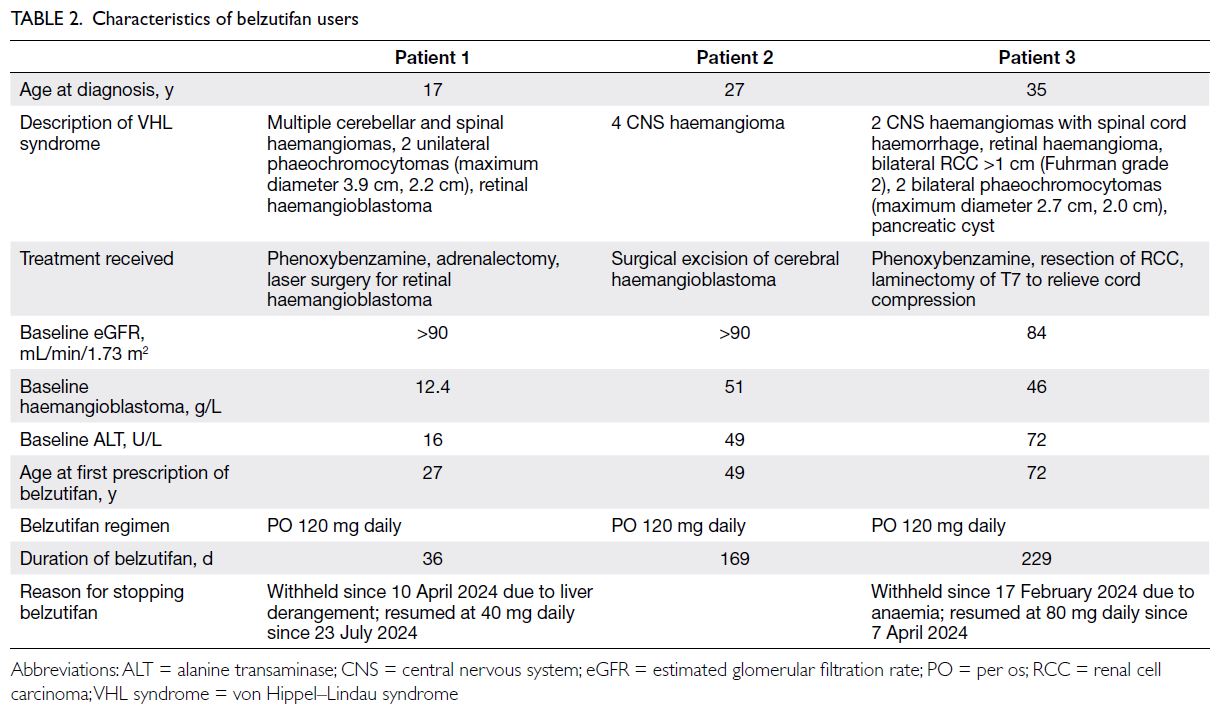
Table 2. Characteristics of belzutifan users
von Hippel–Lindau syndrome–attributable
healthcare costs
During the study period, a total of 368 ED visits, 1209
inpatient admissions, and 5635 days of hospitalisation
were recorded. In total, 21 patients had ICU stays,
amounting to 192 ICU days. These utilisation
patterns translated to an annualised per-patient ED
visit–related cost of HK$8625 and an annualised
per-patient inpatient admission–related cost of
HK$129 968.4 Six patients required dialysis, and 4373
dialysis sessions were performed during the study
period, resulting in a total cost of HK$28.8 million
(HK$6580 per dialysis session).4 For the belzutifan
patient cohort, no ED visits or inpatient admissions
were recorded after initiation of belzutifan therapy,
likely due to the short follow-up duration after
the prescription of this drug newly approved by
the United States Food and Drug Administration.
Consequently, we could not directly compare the
healthcare cost burden between belzutifan users and
non-users.
The pattern of tumour-related surgeries and
accident and emergency admissions in VHL patients
was highly variable; some patients experienced
periods of intense activity followed by quieter
phases, suggesting non-linear disease progression.
Tumour-related operations and deaths since
diagnosis are shown in Figure 3, whereas accident
and emergency admissions are presented in Figure 4,
highlighting individual disease burden. Monitoring
and management should be tailored to address these
fluctuating needs.
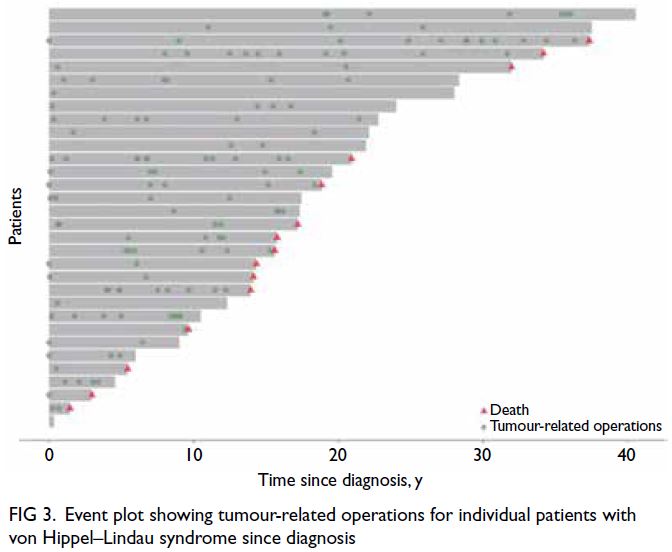
Figure 3. Event plot showing tumour-related operations for individual patients with von Hippel–Lindau syndrome since diagnosis
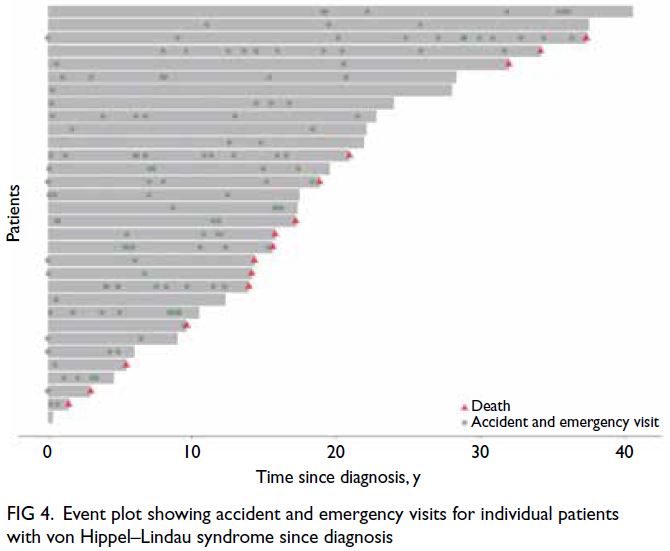
Figure 4. Event plot showing accident and emergency visits for individual patients with von Hippel–Lindau syndrome since diagnosis
Discussion
From this review, we observed that VHL-spectrum
diseases emerged at a young age and recurred
throughout patients’ lives, leading to considerable
morbidity and mortality. This finding is consistent
with existing literature. There is a pressing need
to improve the current care of VHL syndrome in
Hong Kong to enhance patients’ life trajectories and
quality of life.
Pathophysiology
The VHL protein normally functions as an E3
ubiquitin ligase that facilitates ubiquitination of
the alpha subunit of HIF, leading to its proteolysis.5
In VHL patients, genetic alterations reduce VHL
protein activity, thereby disinhibiting HIF-mediated
transcription. Consequently, the overexpression
of vascular endothelial growth factor, cyclin D1,
glucose transporter 1, and erythropoietin promotes
neoplastic growth.5 6 The resultant tissue overgrowth
leads to early-onset, recurrent, and multi-system
benign and malignant neoplasms.1
Functional impairment in patients
Patients with VHL syndrome experience a lifelong
journey with the disease, characterised by substantial
morbidity and mortality.
An Italian study of 128 VHL patients showed
that the natural history varied according to disease
manifestations.7 For RCC, the median age at first
presentation was 31 years,7 similar to our cohort,
which had a median age of 27.4 years. The first
progression typically occurred after 7 to 8 years;
a second progression followed 1 to 2 years later.
von Hippel–Lindau syndrome–related cerebellar
haemangioblastomas generally developed at a median
age of 30 years and progressed relatively consistently
every 3.5 years. The cumulative incidences of
disability were 26.5% for CNS involvement, 16.4%
for visual disturbance, 12.5% for hearing loss, 10.9%
for adrenergic dysfunction, 4.6% for pancreatic
morbidity, and 1.5% for renal impairment.7 One
patient died of metastatic RCC (0.8%), another
entered a vegetative state after a CNS procedure
(0.8%), and five died of postoperative complications (3.9%).7 Overall, the average Karnofsky performance
status was 80% at the end of follow-up.7
In contrast, in our cohort, the nine patients who
died of VHL syndrome–related tumours succumbed
to the disease itself, rather than postoperative
complications. This highlights the substantial impact
of such tumours on patient mortality, underscoring
the need for vigilant monitoring and comprehensive
management strategies to improve outcomes.
Surgery and radiosurgery for von Hippel–Lindau syndrome–related tumours
Central nervous system haemangioblastomas
represent a major and disabling manifestation of
VHL syndrome. A prospective natural history study
focusing on stereotactic radiosurgery for CNS-Hb in
VHL patients reported outcomes from 20 individuals
treated for 44 lesions.8 Most lesions were located in
the cerebellum (n=39), and five in the brainstem.
The mean age at treatment was 37.5 ± 12.0 years.8
All patients were alive at a mean follow-up interval
of 8.5 years. Tumours (mean volume: 0.5 ± 0.7
cm3) were treated with a mean prescription dose of
18.9 Gy (range, 12-24) to the tumour margin, resulting
in local control rates of 91%, 83%, 61%, and 51% at 2, 5,
10, and 15 years, respectively.8 Despite the favourable
early response to stereotactic radiosurgery, VHL
syndrome–related haemangioblastomas tend to
progress during long-term follow-up.
With respect to the treatment of VHL
syndrome–related RCC (ie, VHL-RCC), the rule of
thumb is to strike a balance between oncological
control and renal function preservation to avoid
or delay ESRF. Common strategies include
nephron-sparing surgery and ablative therapies. A
retrospective review of VHL-RCC by Duffey et al9
suggested that 3 cm was a reasonable cutoff, beyond
which metastasis may occur earlier; therefore,
nephron-sparing surgery would be indicated. In a
cohort of 54 VHL patients who underwent nephron-sparing
surgery, nephrectomy, or thermal ablation
for RCC,10 97 kidney treatments were performed.
Nephron-sparing surgery was adopted in 96% of first
and 67% of second interventions. The probabilities
of a second surgery were 21% at 5 years and 42% at
10 years. The overall survival and cancer-specific
survival rates were 82.5% and 90.5%, respectively, at
the 10-year follow-up. No metastasis was observed
for RCCs with a maximum diameter smaller than 4 cm.10
Systemic therapies for von Hippel–Lindau
syndrome
With greater understanding of the genetics and
pathophysiology of VHL syndrome, researchers have
been actively developing effective systemic therapies.
The advent of belzutifan has revolutionised systemic
therapy for VHL syndrome. This HIF-2α inhibitor
demonstrated satisfactory objective response
rates for RCC (49%), pancreatic lesions (77%), and
CNS-Hb (30%), along with an acceptable safety
profile—anaemia and fatigue were the most common
side-effects.2 On 13 August 2021, belzutifan was
approved by the United States Food and Drug
Administration for use in adult VHL patients who
need treatment for associated RCC, CNS-Hb, or
pancreatic neuroendocrine tumours not requiring
immediate surgery.11
The LITESPARK-004 (MK-6482-004) phase
2 study further supports the clinical benefits of
belzutifan in patients with VHL syndrome.12 With
over 2 years of follow-up data, the study demonstrated
sustained efficacy in reducing tumour burden
across multiple organs.12 Objective response rates
were consistent with earlier findings: 49% for RCC,
77% for pancreatic lesions, and 30% for CNS-Hb.12
Notably, the responses were durable, with many
patients experiencing prolonged disease control
without surgical intervention. The safety profile
remained acceptable; anaemia and fatigue were
the most common adverse events.12 These findings
reinforce belzutifan’s potential as a transformative
systemic therapy, offering a non-invasive alternative
to repeated surgeries and improving patient quality
of life. Continued research and access to such
therapies, particularly in Asian populations, are
essential.
Socio-economic impact
von Hippel–Lindau syndrome–related RCC is a
notable malignancy within the disease spectrum.
In our cohort, the annualised per-patient ED visit–related cost for VHL-RCC patients was HK$2070,
and the annualised inpatient admission cost was
HK$23 965. In comparison, an American study
reported that VHL-RCC patients (n=160) incurred
US$36 450 more annually than the control group
(n=800), including US$21 123 more for RCC
management.13 Among complications, ESRF was the most costly, requiring US$65 338 over 6 months
post-nephrectomy.13 Similarly, our cohort incurred
approximately HK$28.8 million during the study
period for repeated dialysis in six patients with ESRF.
Another claims-based study showed that
CNS-Hb and pancreatic neuroendocrine tumours
due to VHL syndrome similarly increased annual
healthcare costs by US$49 645 compared with the
control group.14 These findings underscore the
importance of novel therapies that can alleviate both
clinical and economic burdens.
In our local hospital system, the estimated
annualised per-patient ED visit–related cost
was HK$8625, and the annualised per-patient
inpatient admission–related cost was HK$129 968.
Additionally, dialysis for the six patients with ESRF
required an additional HK$28.8 million. We did
not include calculations for the surgical treatment
of all tumours and related management due to the
practical difficulties of cost estimation within the
public hospital system. Nevertheless, we expect
these costs to be substantial. Although the current
drug cost for belzutifan is high (estimated at around
CAD$17 920 per 28 days15), the medical expenses
associated with the natural course of VHL syndrome
are also considerable. Evidence regarding the
cost-effectiveness of medical therapies, including belzutifan, is still emerging; it is important to
consider the composite outcomes of mortality,
healthcare-related costs, irreversible morbidities,
and social dysfunction. Further economic studies
are warranted to quantify the potential cost savings
associated with this novel treatment.
Future directions to optimise care
von Hippel–Lindau syndrome greatly affects
patients’ clinical outcomes and quality of life.
Frequent hospitalisations, repeated medical and
surgical therapies, and recurrent tumours contribute
to cumulative morbidities and mortality. The need
for multidisciplinary care, ongoing surveillance for
recurrence, and genetic counselling further add
to the disease burden. Thus, VHL patients require
improved access to novel medications.
As our results suggest, the management of VHL
syndrome should be holistic. Patients with multiple
VHL syndrome–related conditions should be
discussed at multidisciplinary meetings to facilitate
treatment prioritisation. A sensible approach
would be to address the most life-threatening and
symptomatic disease first.
The initial local experience of using belzutifan
was promising, with manageable toxicity profiles.
With the advent of its coverage by the Samaritan
Fund for eligible patients,16 the role of belzutifan is
expected to rise in local VHL management. While
its safety and efficacy have been demonstrated in
Western populations, its benefits for Asian patients
remain to be fully defined. This retrospective study
showed that one belzutifan user in the cohort
developed fewer new-onset VHL syndrome–related
conditions than non-users. However, the small
sample size (three belzutifan users among 32 VHL
patients) limits generalisability. Nevertheless, the
encouraging initial results of belzutifan in controlling
tumour growth in the kidneys, CNS, retina, and
pancreas support the need for coordinated efforts in
resource allocation and the establishment of subsidy
schemes.3 With increased use of the medication,
overall healthcare costs are expected to decline
due to reductions in surgeries and hospitalisations.
Given the rarity of VHL syndrome, future clinical
trials should ideally be multi-national and multi-centre.
Local registries should also be established
to facilitate long-term follow-up, clinical trial
enrolment, and policy development for this patient
group. Additionally, patient support groups, social
support initiatives, and increased attention to
psychological well-being would help provide holistic
care for VHL patients. Addressing the financial and
disease-related burdens faced by this vulnerable
population is essential to improving their quality of
life and long-term outcomes.
Limitations
Our cohort did not include all VHL patients in Hong
Kong. Assuming an incidence of one in 27 30017 and
a total population of 7 million in Hong Kong,18 the
estimated number of VHL patients in this locality
is approximately 250, excluding those who did not
present to the participating hospitals or whose
follow-up data were unavailable. Nevertheless, our
study offers the first insight into the clinical journey
of local VHL patients.
Conclusion
Overall, VHL patients experience early-onset and
recurrent multi-systemic illness, with a substantial
risk of irreversible morbidity and mortality.
Multidisciplinary care and the promotion of
effective treatments such as belzutifan may improve
the management of this rare but important disease.
Author contributions
Concept or design: AYH Lee, DKW Leung.
Acquisition of data: CH Leung, KHY Tsang, A Yiu, CYK Ho.
Analysis or interpretation of data: AYH Lee, DKW Leung, CH Leung.
Drafting of the manuscript: AYH Lee, DKW Leung.
Critical revision of the manuscript for important intellectual content: JMK Ho, CF Ng.
Acquisition of data: CH Leung, KHY Tsang, A Yiu, CYK Ho.
Analysis or interpretation of data: AYH Lee, DKW Leung, CH Leung.
Drafting of the manuscript: AYH Lee, DKW Leung.
Critical revision of the manuscript for important intellectual content: JMK Ho, CF Ng.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
As an editor of the journal, CF Ng was not involved in the peer review process. Other authors have disclosed no conflicts of interest.
Acknowledgement
The authors thank the following contributors for their
expertise and support in the research: Dr Jeffrey SK Chan
and Dr Esther TW Cheng of the Cardio-Oncology Research
Unit, Cardiovascular Analytics Group, Hong Kong, China–UK Collaboration; and Dr Brian WH Siu, Dr Ivan CH Ko, Dr
Chris HM Wong, and Dr Alex Liu of the Division of Urology,
Department of Surgery, Faculty of Medicine, The Chinese
University of Hong Kong.
Funding/support
This research received no specific grant from any funding
agency in the public, commercial, or not-for-profit sectors.
Ethics approval
This research was approved by the Joint Chinese University of
Hong Kong–New Territories East Cluster Clinical Research
Ethics Committee, Hong Kong (Ref No.: 2024.435). A waiver
of patient consent was granted by the Committee due to the
retrospective nature of the research.
References
1. Couch V, Lindor NM, Karnes PS, Michels VV. von Hippel–Lindau disease. Mayo Clin Proc 2000;75:265-72. Crossref
2. Binderup ML, Galanakis M, Budtz-Jørgensen E,
Kosteljanetz M, Luise Bisgaard M. Prevalence, birth
incidence, and penetrance of von Hippel–Lindau disease
(vHL) in Denmark. Eur J Hum Genet 2017;25:301-7. Crossref
3. Jonasch E, Donskov F, Iliopoulos O, et al. Belzutifan for
renal cell carcinoma in von Hippel–Lindau disease. N Engl
J Med 2021;385:2036-46. Crossref
4. Hospital Authority, Hong Kong. Hospital Authority
Annual Report 2007-2008. Available from: https://www.ha.org.hk/ho/corpcomm/Annual%20Report/2007-08.pdf. Accessed 10 Oct 2024.
5. Choueiri TK, Kaelin WG Jr. Targeting the HIF2-VEGF axis
in renal cell carcinoma. Nat Med 2020;26:1519-30. Crossref
6. Haase VH. The VHL tumor suppressor: master regulator of
HIF. Curr Pharm Des 2009;15:3895-903. Crossref
7. Feletti A, Anglani M, Scarpa B, et al. von Hippel–Lindau
disease: an evaluation of natural history and functional
disability. Neuro Oncol 2016;18:1011-20. Crossref
8. Asthagiri AR, Mehta GU, Zach L, et al. Prospective
evaluation of radiosurgery for hemangioblastomas in von
Hippel–Lindau disease. Neuro Oncol 2010;12:80-6. Crossref
9. Duffey BG, Choyke PL, Glenn G, et al. The relationship
between renal tumor size and metastases in patients with
von Hippel–Lindau disease. J Urol 2004;172:63-5. Crossref
10. Jilg CA, Neumann HP, Gläsker S, et al. Nephron sparing
surgery in von Hippel–Lindau associated renal cell
carcinoma; clinicopathological long-term follow-up. Fam
Cancer 2012;11:387-94. Crossref
11. United States Food and Drug Administration. FDA
approves belzutifan for cancers associated with von
Hippel–Lindau disease. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-belzutifan-cancers-associated-von-hippel-lindau-disease. Accessed 6 Aug 2025.
12. Jonasch E, Iliopoulos O, Kimryn Rathmell W, et al.
LITESPARK-004 (MK-6482-004) phase 2 study of
belzutifan, an oral hypoxia-inducible factor 2α inhibitor
(HIF-2α), for von Hippel–Lindau (VHL) disease: update
with more than two years of follow-up data. J Clin Oncol
2022;40 (Suppl):4546. Crossref
13. Jonasch E, Song Y, Freimark J, et al. Epidemiology
and economic burden of von Hippel–Lindau disease–associated renal cell carcinoma in the United States. Clin
Genitourin Cancer 2023;21:238-47. Crossref
14. Jonasch E, Song Y, Freimark J, et al. Epidemiology
and economic burden of von Hippel–Lindau disease–associated central nervous system hemangioblastomas and
pancreatic neuroendocrine tumors in the United States.
Orphanet J Rare Dis 2024;19:73. Crossref
15. Belzutifan (Welireg): CADTH Reimbursement Review:
Therapeutic area: von Hippel–Lindau disease–associated
tumours [Internet]. Ottawa (ON): Canadian Agency
for Drugs and Technologies in Health; 2023 Nov.
Pharmacoeconomic Review. Available from: https://www.ncbi.nlm.nih.gov/books/NBK599999/. Accessed 10 Oct 2024.
16. Samaritan Fund. Items supported by the Samaritan Fund.
Available from: https://www.ha.org.hk/haho/ho/sf/SF_Items_en.pdf. Accessed 18 Aug 2025.
17. Rare Disease Hong Kong. von Hippel–Lindau Disease.
About Rare Diseases Rare Disease Wiki. Available from:
https://rdhk.org/post/data?mid=15&id=13471&lang=en. Accessed 10 Oct 2024.
18. Census and Statistics Department, Hong Kong SAR
Government. Year-end Population for 2023 [20 Feb 2024].
Available from: https://www.censtatd.gov.hk/en/press_release_detail.html?id=5386. Accessed 10 Oct 2024.