Hong Kong Med J 2024 Jun;30(3):241–4 | Epub 31 May 2024
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Hypokalaemic hypertension and 17-alpha-hydroxylase/17,20-lyase deficiency in a young girl: a case report
HN Yau, FHKCPaed, FHKAM (Paediatrics)1; WC Lo, FHKCPaed, FHKAM (Paediatrics)2; YP Yuen, FHKAM (Pathology), FRCPA3; MT Leung, FHKCPath, FHKAM (Pathology)4; KL Ng, FHKAM (Paediatrics), FRCPCH2
1 Department of Paediatrics and Adolescent Medicine, Tuen Mun Hospital, Hong Kong SAR, China
2 Department of Paediatrics and Adolescent Medicine, United Christian Hospital, Hong Kong SAR, China
3 Department of Pathology, Hong Kong Children’s Hospital, Hong Kong SAR, China
4 Department of Pathology, Queen Elizabeth Hospital, Hong Kong SAR, China
Corresponding author: Dr HN Yau (nicoleyau@fellow.hkam.hk)
 Full paper in PDF
Full paper in PDF
Case presentation
A young girl aged 9 years 6 months was admitted
to hospital in July 2022 with coronavirus disease
2019 infection and found to have refractory
hypokalaemia. She reported good past health and
denied taking any supplements or medications.
Her nonconsanguineous parents and 4-year-old
younger brother were healthy and family history was
unremarkable.
Her body weight and height were 35.8 kg at
the 75th to 95th percentile and 140 cm at the 75th
percentile, respectively. Her body mass index was
18.3 kg/m2 at the 75th percentile. Blood pressure was
noted to be persistently high up to 152/117 mm Hg
(112/73 mm Hg being the 90th percentile for her
gender, age and height according to the American
Academy of Pediatrics1). Cardiovascular and
abdominal examinations were unremarkable. There
was no hyperpigmentation or sign of virilisation.
She was prepubertal, with normal female external
genitalia.
Ambulatory blood pressure monitoring
confirmed stage 2 hypertension. Electrolytes and
hormone profile are summarised in the Table.
Her bone age was 8 years 10 months according to
the Greulich and Pyle method of assessment. A
standard-dose Synacthen test did not stimulate a
rise in 17-hydroxyprogesterone or cortisol level.
Ultrasound of the pelvis revealed prepubertal uterus
and bilateral ovaries. Her karyotype was 46,XX.
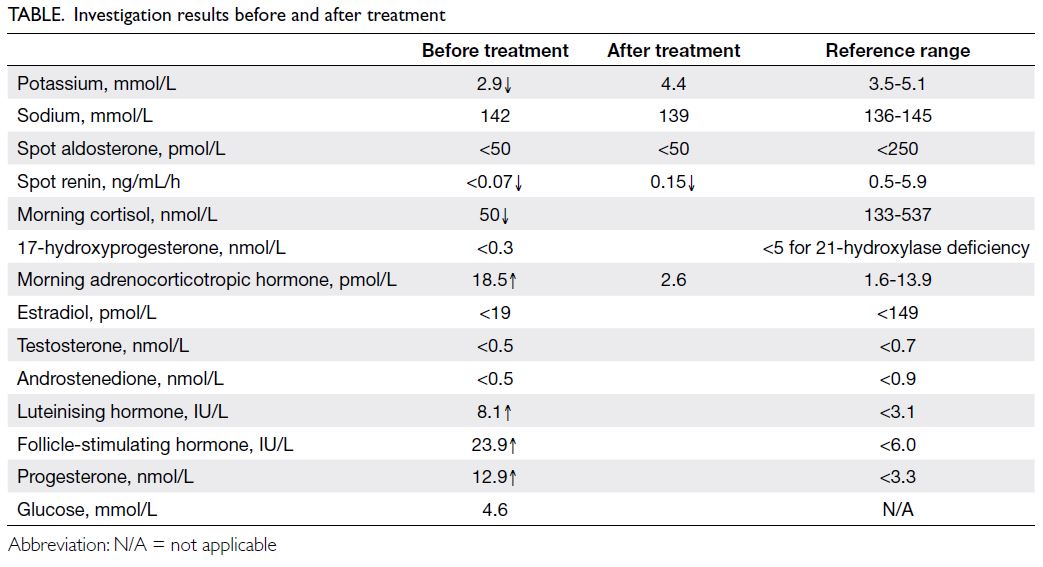
Table. Investigation results before and after treatment
Urine steroid profile showed a
characteristic pattern compatible with 17-alpha-hydroxylase/17,20-lyase deficiency (17OHD), with
a lack of androgen metabolites and an excess of
progesterone, pregnenolone, and corticosterone
metabolites (online supplementary Appendix).
Oral hydrocortisone was commenced at 7.6
mg/m2/day. Blood pressure improved to 119/81
mm Hg after 2 weeks. Spironolactone was added for
better control.
Genetic analysis revealed compound heterozygous pathogenic variants in the CYP17A1 gene: c.297+2T>C in intron 1 and c.849delC
p.(Ser284Glnfs*13) in exon 5, which confirmed the
diagnosis of 17OHD. Parental testing confirmed that
the two CYP17A1 variants were in trans, and the
younger brother was a carrier.
Discussion
17-alpha-hydroxylase/17,20-lyase deficiency is a
rare type of congenital adrenal hyperplasia that
accounts for 1% of cases with an estimated incidence
of 1 in 50 000 to 100 000.2 17-alpha-hydroxylase
and 17,20-lyase enzyme defects result in cortisol
and sex hormone deficiency, with compensatory
rise in adrenocorticotropic hormone that drives
excessive production of 11-deoxycorticosterone and
corticosterone (Fig).3
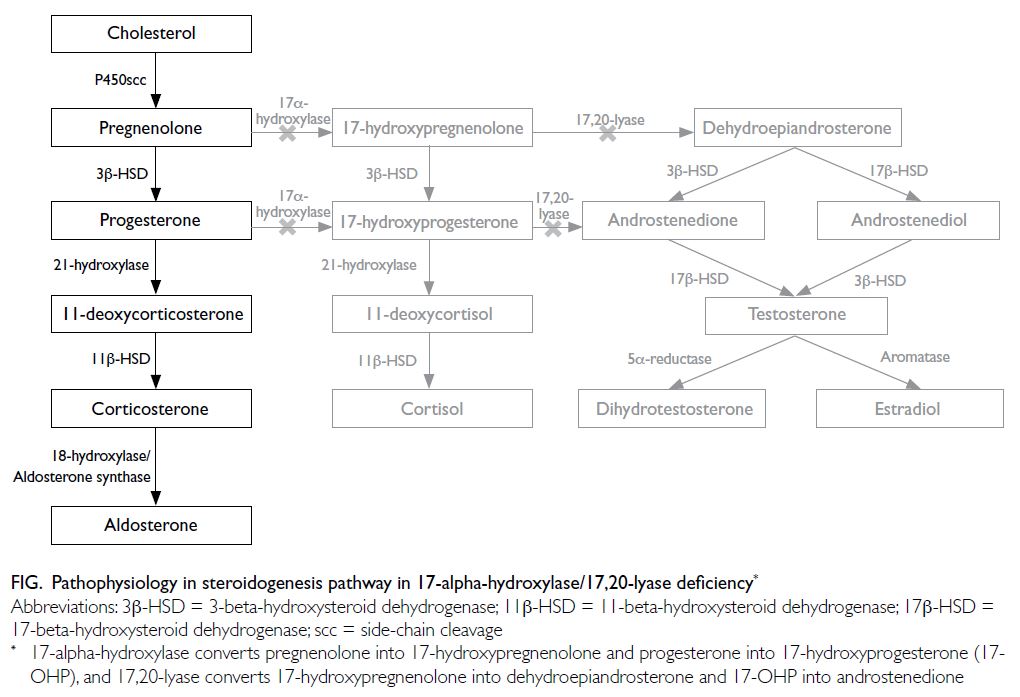
Figure. Pathophysiology in steroidogenesis pathway in 17-alpha-hydroxylase/17,20-lyase deficiency
Unlike the pathophysiology of the more
common types of congenital adrenal hyperplasia
such as 21-hydroxylase deficiency and 11-beta-hydroxylase
deficiency, 17OHD does not cause
excessive testosterone production with consequent
virilisation in affected 46,XX females or precocious
puberty in both sexes.3 Instead, due to the lack
of sex steroid hormone production, it results in
undervirilisation in 46,XY males and sexual infantilism
in 46,XX females.3 The excessive accumulation
of deoxycorticosterone and corticosterone exerts
potent mineralocorticoid effects, resulting in
sodium and fluid retention, hence suppression of
renin with hypokalaemic hypertension,2 as in our
patient. A high concentration of corticosterone
provides sufficient glucocorticoid effect to prevent
adrenal crisis.3 The classic presentation of 17OHD is
a phenotypic female with delayed puberty, primary
amenorrhoea, and hypokalaemic hypertension
with diagnosis often made in young adulthood.2 4 5
Cases of retained partial enzyme activity resulting
in ambiguous genitalia in 46,XY males have been
reported.3 4
Delayed diagnosis of 17OHD is not uncommon due to its subtle and late presentation. Hypokalaemic
hypertension should prompt a clinician to search
for secondary causes of hypertension. Plasma renin
activity is an important investigation to differentiate
the causes, and would be suppressed in 17OHD by
the potent mineralocorticoid activity, and is high in renovascular disease.6 On the contrary, aldosterone
level could be suppressed, normal or raised in
17OHD.2 4 5 Low aldosterone level arises as a result
of a suppressed renin angiotensin system while
high level might be related to more severe enzyme
defects resulting in greater production of end product from aldosterone precursors.4 As 17OHD
has a characteristic pattern of metabolite excretion
and metabolite ratios on urine steroid profiling,
this profiling is an important investigation when
diagnosing the condition.
There is no consensus guideline on the
management of 17OHD. The mainstay of treatment
is glucocorticoid and sex hormone replacement.
The use of glucocorticoid would decrease the
adrenocorticotropic hormone drive and production
of deoxycorticosterone and corticosterone, which
would facilitate improved blood pressure control and
electrolyte balance.2 Different forms and dosages of
glucocorticoid replacement have been described in
the literature, ranging from dexamethasone 0.25 mg
to 1 mg daily, or equivalent.2 4 5 In some cases, an
antihypertensive agent with a mineralocorticoid
antagonist effect such as spironolactone or
eplerenone may be required for blood pressure
control.3 5 Patients may develop end organ damage
such as hypertensive retinopathy if blood pressure
control is suboptimal.2 Deoxycorticosterone and
corticosterone levels might not be normalised
despite treatment. Blood pressure control, electrolyte
balance, and renin level are more important markers
of disease control.4
Our patient was phenotypically female, in
line with her genotypic sex. Her hormone blood
test revealed hypergonadotropic hypogonadism
due to lack of sex hormone production. Oestrogen
and progestin replacement should be commenced
at an appropriate time during adolescence, or upon
diagnosis in adulthood, to induce secondary sexual
characteristics and cyclic uterine bleeding.5 Sex
steroid replacement therapy may improve bone
mineral density in 17OHD patients and prevent
osteoporosis. For genotypic males, psychological
assessment should determine gender preference
prior to initiation of sex hormone replacement,
and intraabdominal testes should be removed to
prevent malignant change.3 A multidisciplinary
team approach involving an endocrinologist,
surgeon, psychiatrist, psychologist and social worker
is essential and can help in parental counselling,
achieving family consensus for gender assignment,
and formulation of an individualised plan for each
patient.
Genetic test on CYP17A1 is important to
make the diagnosis of 17OHD. To date, the Human
Gene Mutation Database has reported >100
different types of mutations on the CYP17A1 gene.7
Genetic analysis showed compound heterozygous
pathogenic variants in the CYP17A1 gene (reference
transcript: NM_000102.4): c.297+2T>C in intron
1 and c.849delC p.(Ser284Glnfs*13) in exon 5.
c.297+2T>C is a splice site variant that has been
previously described in patients with 17OHD
(ClinVar accession No.: VCV0004319808), while c.849delC is a truncating variant that creates a
premature termination codon. It is expected to result
in an absent or disrupted protein product (ClinVar
accession No.: VCV0014174348). This variant has an
extremely low minor allele frequency in the general
population and has not been reported in patients
with CYP17A1-related disease.
17-alpha-hydroxylase/17,20-lyase deficiency
is associated with infertility due to premature
follicular arrest and poor endometrial development
for implantation.9 Women with 17OHD have
difficulty conceiving, even with the help of assisted
reproductive technology.9 Though not promising,
fertility had been possible for patients with partial
17OHD. Patients should be counselled about
potential fertility difficulties and referred to an
assisted reproductive service when appropriate.
To conclude, 17OHD should be considered in
a young hypertensive individual with hypokalaemia.
Timely management with glucocorticoid and sex
hormone replacement can ameliorate the morbidity
of hypertension and hypogonadism. Multidisciplinary
collaboration is advised, especially for patients with
gender identification or infertility issues.
Author contributions
All authors contributed to the concept or design, acquisition
of data, analysis or interpretation of data, drafting of the
manuscript, and critical revision of the manuscript for
important intellectual content. All authors had full access to
the data, contributed to the study, approved the final version
for publication, and take responsibility for its accuracy and
integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of
Helsinki. The parents of the patient provided written consent
for publication of this case report.
Supplementary material
The supplementary material was provided by the authors
and some information may not have been peer reviewed.
Accepted supplementary material will be published as
submitted by the authors, without any editing or formatting.
Any opinions or recommendations discussed are solely those
of the author(s) and are not endorsed by the Hong Kong
Academy of Medicine or the Hong Kong Medical Association.
The Hong Kong Academy of Medicine and the Hong Kong
Medical Association disclaim all liability and responsibility
arising from any reliance placed on the content.
References
1. Flynn JT, Kaelber DC, Baker-Smith CM, et al. Clinical
practice guideline for screening and management of high
blood pressure in children and adolescents. Pediatrics
2017;140:e20171904. Crossref
2. Kim SM, Rhee JH. A case of 17 alpha-hydroxylase
deficiency. Clin Exp Reprod Med 2015;42:72-6. Crossref
3. Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase
deficiencies, genetic and pharmacologic. J Steroid Biochem
Mol Biol 2017;165(Pt A):71-8. Crossref
4. Peter M, Sippell WG, Wernze H. Diagnosis and treatment
of 17-hydroxylase deficiency. J Steroid Biochem Mol Biol
1993;45:107-16. Crossref
5. Han LH, Wang L, Wu XY. 17 alpha-hydroxylase deficiency: a case report of young Chinese woman with a rare gene mutation. Clin Case Rep 2022;10:e6109. Crossref
6. Choi KB. Hypertensive hypokalemic disorders. Electrolytes
Blood Press 2007;5:34-41. Crossref
7. Institute of Medical Genetics in Cardiff. Human Gene
Mutation Database. Available from: https://www.hgmd.cf.ac.uk/ac/index.php. Accessed 24 May 2024.
8. National Center for Biotechnology Information, National
Library of Medicine. ClinVar. Available from: https://www.ncbi.nlm.nih.gov/clinvar/". Accessed 24 May 2024.
9. Marsh CA, Auchus RJ. Fertility in patients with genetic
deficiencies of cytochrome P450c17 (CYP17A1): combined
17-hydroxylase/17,20-lyase deficiency and isolated 17,20-lyase deficiency. Fertil Steril 2014;101:317-22.Crossref