Hong Kong Med J 2023 Oct;29(5):462–5 | Epub 26 Jul 2023
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Occult intravascular large B-cell lymphoma presenting as postoperative thrombotic microangiopathy: a case report
WK Lam, MB, BS1; Edmond SK Ma, FHKCPath, MD2; SY Kong, FHKCP, FHKAM (Medicine)3; SF Yip, FHKCPath, FHKCP1
1 Department of Clinical Pathology, Tuen Mun Hospital, Hong Kong SAR, China
2 Clinical Laboratory, Hong Kong Sanatorium & Hospital, Hong Kong SAR, China
3 Department of Medicine and Geriatrics, Pok Oi Hospital, Hong Kong SAR, China
Corresponding author: Dr WK Lam (lwk936@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Case presentation
In August 2017, a 59-year-old male with good past
health was admitted via the emergency department
with a 4-day history of abdominal distension and
epigastric pain. He had passed no stool for 2 days.
Physical examination revealed epigastric tenderness
and an empty rectum. Abdominal X-ray showed
intestinal obstruction and erect chest X-ray showed
no free gas under the diaphragm. Blood tests on
admission revealed a normal complete blood count
and acute renal failure (Table). The patient was
started on intravenous (IV) fluid and IV amoxicillin
and clavulanate. Urgent computed tomography (CT)
abdomen and pelvis with contrast showed acute
appendicitis with perforation and dilated small bowel
without an obvious transition point. Emergency
laparotomy revealed appendiceal diverticulitis with
walled-off localised abscess formation around the
appendix. The base of the appendix was healthy.
Microscopic examination of the appendix showed
dense mixed inflammatory cell infiltration in
periappendiceal fat and serositis, with no evidence
of malignancy, thrombosis or ischaemia.
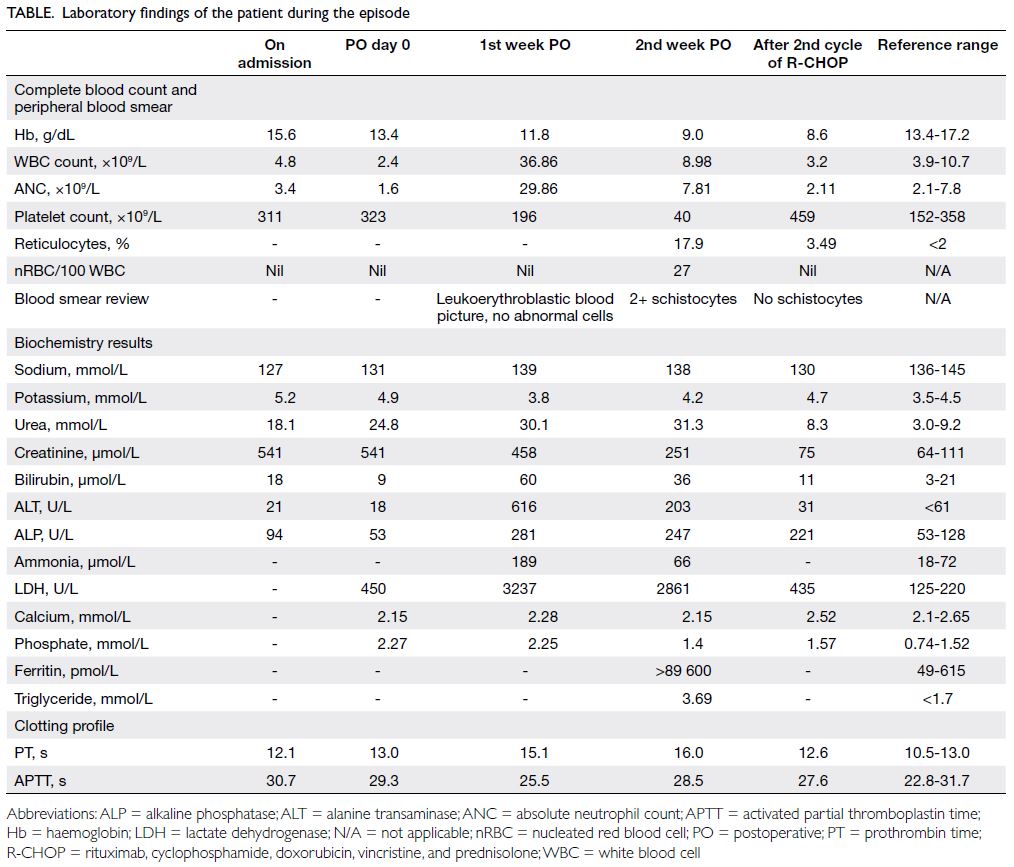
Table. Laboratory findings of the patient during the episode
Three days postoperatively, the patient
developed fever and hypotension and was given
IV piperacillin and tazobactam. There was no
organomegaly, lymphadenopathy or skin lesions on
physical examination. He became dull looking but
there was no focal neurological deficit. Septic workups
with blood and urine were negative. Sputum culture
showed Enterobacter cloacae, resistant to amoxicillin
and clavulanate. Widal test, Weil–Felix test, human
immunodeficiency virus serology and tests for viral
hepatitis were all negative. A second abdomen
CT showed no gross infective foci and only a few
subcentimetre lymph nodes. Plain brain CT showed
no focal intracranial lesions. Blood tests showed a
leukoerythroblastic blood picture and no circulating
abnormal cells, and acute hepatic and renal failure
with markedly elevated lactate dehydrogenase
level (Table). Fever did not abate despite IV
piperacillin and tazobactam, IV meropenem and IV anidulafungin were prescribed for support. In the
second week postoperatively, blood tests showed
progressive anaemia and thrombocytopenia with
microangiopathic haemolytic anaemia (MAHA)
and a leukoerythroblastic blood picture (Table and
Fig a). Ferritin and triglyceride levels were markedly elevated.
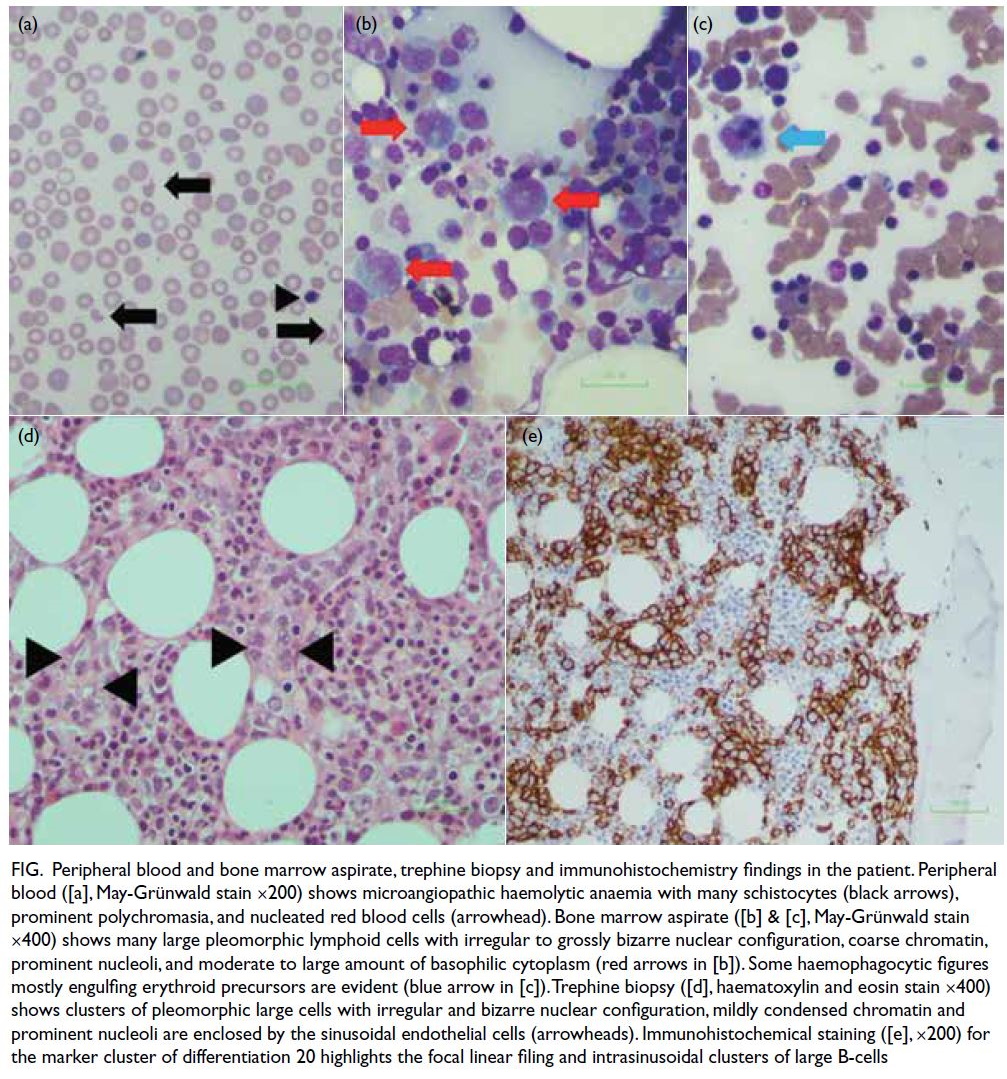
Figure. Peripheral blood and bone marrow aspirate, trephine biopsy and immunohistochemistry findings in the patient. Peripheral blood ([a], May-Grünwald stain ×200) shows microangiopathic haemolytic anaemia with many schistocytes (black arrows), prominent polychromasia, and nucleated red blood cells (arrowhead). Bone marrow aspirate ([b] & [c], May-Grünwald stain ×400) shows many large pleomorphic lymphoid cells with irregular to grossly bizarre nuclear configuration, coarse chromatin, prominent nucleoli, and moderate to large amount of basophilic cytoplasm (red arrows in [b]). Some haemophagocytic figures mostly engulfing erythroid precursors are evident (blue arrow in [c]). Trephine biopsy ([d], haematoxylin and eosin stain ×400) shows clusters of pleomorphic large cells with irregular and bizarre nuclear configuration, mildly condensed chromatin and prominent nucleoli are enclosed by the sinusoidal endothelial cells (arrowheads). Immunohistochemical staining ([e], ×200) for the marker cluster of differentiation 20 highlights the focal linear filing and intrasinusoidal clusters of large B-cells
Bone marrow biopsy was subsequently
performed and revealed many abnormal
pleomorphic large cells and haemophagocytosis
(Fig). In view of the fever of unknown origin,
cytopenia, hypertriglyceridaemia, markedly elevated
ferritin and haemophagocytosis in the bone marrow,
a diagnosis was reached of haemophagocytic
lymphohistiocytosis (HLH) and dexamethasone
15 mg daily orally and IV immunoglobulin
started. Trephine biopsy showed prominent
infiltration of pleomorphic large cells in focal
linear profiling and intrasinusoidal clusters. With
immunohistochemistry, the large cells were shown to
be positive for the markers cluster of differentiation
(CD20) [Fig e], CD30, CD5, and negative for activin
receptor-like kinase 1 and cyclin D1. Epstein-Barr
virus–encoded small RNA in situ hybridisation was
negative. A diagnosis was made of intravascular
large B-cell lymphoma (IVLBCL).
The ADAMTS13 (a disintegrin and
metalloproteinase with a thrombospondin type 1
motif, member 13) activity, antigen and autoantibody
assays were performed. There was severe reduction in
ADAMTS13 activity to 5% with ADAMTS13 antigen
level moderately reduced to 215 ng/mL (reference
range: 430-970) and autoantibody level at 2.5 units/mL (negative: <15). This confirmed ADAMTS13
deficiency and functional defect compatible with
the picture of malignancy-associated thrombotic
thrombocytopenic purpura (TTP). The patient was
prescribed immunochemotherapy with rituximab
(recombinant anti-CD20), cyclophosphamide,
vincristine and prednisolone, and rasburicase
for prophylaxis of tumour lysis syndrome.
Hydroxydaunorubicin was added in the second cycle of immunochemotherapy (R-CHOP, ie, a
combination of rituximab, cyclophosphamide,
doxorubicin, vincristine, and prednisolone) after
which blood counts and liver and renal function test
results markedly improved (Table). The patient was
in complete remission after six cycles of R-CHOP.
Nonetheless he died of inoperable squamous cell
carcinoma of oesophagus 3 years after the diagnosis
of lymphoma.
Discussion
Intravascular large B-cell lymphoma is a rare subtype
of extranodal large B-cell lymphoma characterised
by selective growth of neoplastic cells inside the
lumina of small- and medium-sized vessels, often
with an aggressive clinical course. There are two major patterns of clinical presentation: the classic
form with neurocutaneous involvement and the
haemophagocytic syndrome–associated form
with multiorgan failure, hepatosplenomegaly, and
pancytopenia. Diagnosis of IVLBCL is challenging
due to its variable presentation. Imaging may be
negative due to the lack of detectable tumour masses.
The initial presentation of this patient
was atypical, with appendiceal diverticulitis and
perforation. Review of the appendix specimen
confirmed the absence of lymphoma involvement.
There was no hepatosplenomegaly. The abrupt onset
of anaemia and thrombocytopenia with MAHA
raised the possibility of postoperative TTP. Patients
with postoperative TTP characteristically have a
normal complete blood count prior to surgery, but
subsequently show MAHA with thrombocytopenia about 5 to 9 days after surgery.1 Fever, renal
impairment, and neurological symptoms are
variably present. Nonetheless the abundance of
schistocytes and nucleated red cells in the blood
film and acute liver failure did not support a
diagnosis of postoperative TTP, warranting further
investigations.
Neoplastic cells may cause endothelial damage
and result in release of ultra-large von Willebrand
factor multimers. Autoantibodies against
ADAMTS13 may also play a role in pathogenesis,
leading to platelet activation and thrombotic
microangiopathy.2 In our case, ADAMTS13 activity
was markedly reduced at 5%, unusually low for malignancy-associated TTP (median ADAMTS13 activity: 50%).3 Some secondary TTP cases have been reported with markedly reduced ADAMTS13
activity4 but the significance is uncertain. The
ADAMTS13 activity was also disproportionately
lower than the antigen level, indicating a functional
defect that may be seen in acquired TTP. Negative
autoantibody against ADAMTS13 suggests against
a diagnosis of idiopathic TTP. Distinguishing
malignancy-associated TTP from idiopathic or
postoperative TTP is important since therapeutic
plasma exchange is effective in idiopathic or
postoperative TTP but not in malignancy-associated
TTP.
Although the anaemia and thrombocytopenia
could be explained by the MAHA, hyperferritinaemia,
hypertriglyceridaemia and haemophagocytosis in
bone marrow were compatible with HLH. Of note, the
current diagnostic criteria for HLH were originally
proposed for diagnosis in paediatric patients.5 Criteria
cut-offs such as a ferritin level >500 ng/mL may not
be applicable in adults where there are many other
reasons for such a high level. Bone marrow biopsy is
the preferred investigation in the diagnosis of HLH
and IVLBCL. A high index of suspicion should be
maintained since the peripheral blood may not show
abnormalities specific to these diagnoses.
Author contributions
Concept or design: All authors.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: WK Lam.
Critical revision of the manuscript for important intellectual content: WK Lam, ESK Ma, SF Yip.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: WK Lam.
Critical revision of the manuscript for important intellectual content: WK Lam, ESK Ma, SF Yip.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of Helsinki and has provided informed consent for all treatments
and procedures, and consent for publication.
References
1. Eskazan AE, Buyuktas D, Soysal T. Postoperative thrombotic thrombocytopenic purpura. Surg Today 2015;45:8-16. Crossref
2. Sill H, Höfler G, Kaufmann P, et al. Angiotropic large cell lymphoma presenting as thrombotic microangiopathy (thrombotic thrombocytopenic purpura). Cancer 1995;75:1167-70. Crossref
3. George JN. Systemic malignancies as a cause of unexpected microangiopathic hemolytic anemia and thrombocytopenia. Oncology (Williston Park) 2011;25:908-14.
4. Hassan S, Westwood JP, Ellis D, et al. The utility of ADAMTS13 in differentiating TTP from other acute thrombotic microangiopathies: results from the UK TTP Registry. Br J Haematol 2015;171:830-5. Crossref
5. Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124-31. Crossref