© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Importance of cascade family screening and
precision medicine for patients with familial
hyperkalaemia: a case report
HY Lam, MB, BS1; Eugene YH Chan, MRCPCH (UK), FHKAM (Paediatrics)2; Joanna YL Tung, MB, BS, MRCPCH (UK)2; Samantha LK Lee, MB, BS, MPH (HKU)2 Jasmine LF Fung, BBiomedSc3; Mianne Lee, MSc3; Brian HY Chung, MD2,3; Alison LT Ma, MRCPCH (UK), FRCPCH (UK)2
1 Department of Paediatrics and Adolescent Medicine, Alice Ho Miu Ling Nethersole Hospital, Hong Kong
2 Department of Paediatrics and Adolescent Medicine, Hong Kong Children’s Hospital, Hong Kong
3 Department of Paediatrics and Adolescent Medicine, The University of Hong Kong, Hong Kong
Corresponding author: Ms HY Lam (charlottelamhy@gmail.com)
 Full paper in PDF
Full paper in PDF
Case report
The patient was the only child of a non-consanguineous
Chinese couple and was born at
full term by normal vaginal delivery with a birth
weight of 2.85 kg. She had good past health except
for admission for a reflex-anoxic attack at age
7 months. Physical examination at that time was
unremarkable with normal growth parameters and
normal blood pressure for her age. Investigations
showed normal anion gap metabolic acidosis and
hyperkalaemia with a potassium of 7.1 mmol/L but
normal serum sodium and renal function. In view of
the biochemical findings, the provisional diagnosis
was type IV renal tubular acidosis. Fludrocortisone
was started with sodium bicarbonate and furosemide
and a low potassium diet commenced. However,
recurrent episodes of hyperkalaemia continued
despite good compliance, requiring potassium
binder and medication adjustment.
In December 2019, because of the suboptimal
control of hyperkalaemia, with a fluctuating
potassium level of 4.5 to 6.4 mmol/L, the patient
was referred to our unit at age 10 years. Her
blood pressure remained normal throughout.
Blood test results revealed hyperreninaemic
hyperaldosteronism with elevated supine renin
at 5.59 ng/mL/hr (reference: 0.15-2.33 ng/mL/hr)
and elevated aldosterone at 785 pmol/L (reference:
28-444 pmol/L). Pseudohypoaldosteronism type I
was suspected initially.
Trio whole exome sequencing revealed a
paternally inherited novel missense change in
WNK4:NM_032387.5:c.1685A>G:p.(Glu562Gly),
confirming a diagnosis of pseudohypoaldosteronism
type IIB. This patient was coincidentally part of a larger
whole exome sequencing cohort where the study was
focused on cost-effectiveness of rapid whole exome
sequencing.1 The patient was treated with oral 25 mg
hydrochlorothiazide once daily and remained stable
with normal growth and development. The serum potassium and blood pressure were well controlled
and there were no dental or renal complications on
follow-up examination.
Family cascade screening revealed that
her father, paternal grandmother, and paternal
aunt had hyperkalaemia. All were subsequently
confirmed to have the same mutation (Fig 1).
Retrospectively, the father had provided a history
of kidney stone, hyperkalaemia, hypercalciuria and
hypertension since age 35 years. He was found to
have a serum potassium level of 7 mmol/L during
the cascade screening. Although asymptomatic, his
electrocardiogram already showed hyperkalaemic
changes with tented T waves and high blood
pressure at 176/116 mmHg. He was started on
hydrochlorothiazide with normalisation of both
serum potassium and blood pressure a few days
later. The paternal grandmother also had a history of
hyperkalaemia and partially controlled hypertension.
The paternal aunt was asymptomatic with good
past health but was found to be hypertensive and
hyperkalaemic on screening. They were also started
on hydrochlorothiazide with good response.
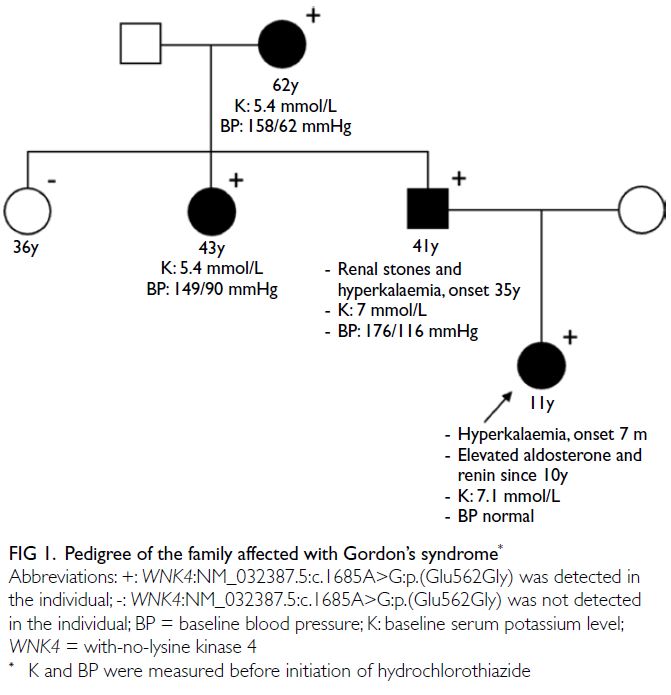
Figure 1. Pedigree of the family affected with Gordon’s syndrome
Discussion
We report a Chinese family with the rare cause of hyperkalaemia and type IV renal tubular acidosis
due to an autosomal dominant condition called
Pseudohypoaldosteronism type II (PHAII). Whole
exome sequencing established the diagnosis in our
index patient, and subsequently, other affected
family members were identified through cascade
family screening.
Pseudohypoaldosteronism type II, also
referred to as familial hyperkalaemic hypertension,
or Gordon’s syndrome, was first described in 1964.2
By 2013, around 100 to 200 individuals and families
with PHAII had been reported.3 Hyperkalaemia with
normal renal function is the main characteristic.
Other common features include metabolic acidosis, diminished renal potassium excretion,
hypercalciuria, low plasma renin/aldosterone
levels and hypertension. Short stature and dental
abnormalities have also been reported.4
Mutation in the gene encoding with-no-lysine
kinase 4 (WNK4), one of the novel proteins
involved in the pathogenesis of PHAII, was
identified in our patient. Other identified genes
include WNK1, kelch-like 3 and cullin 3.3 The
WNK4 is one of the WNK isoforms that regulates
sodium-chloride co-transporter (NCC) in the distal
convoluted tubule. Without the inhibitory effect of
normal WNK4, sodium and chloride reabsorption
are increased via the NCC, leading to volume
expansion and consequent hypertension. The NCC
is more proximal to the epithelial sodium channel
at the distal convoluted tubule. Excessive sodium
reabsorption at NCC results in reduced sodium
delivery to epithelial sodium channels for potassium
secretion at Na+/K+ exchange.3 This theory explains
why thiazide is effective in PHAII as it inhibits NCC.
Increased intracellular sodium due to increased NCC
activity will cause decreased basolateral Na+/Ca2+
exchange that may result in hypercalciuria as in the
father of our index patient. It has been suggested
that mutant WNK4 decreases the activity of renal
outer medullary potassium channels more than
wild-type WNK4. Another proposed mechanism of
this Chloride Shunt Syndrome is increased chloride
reabsorption through paracellular pathways due to
the mutant WNK4, resulting in reduced negativity
at the distal epithelial sodium channel.3 These can
all lead to reduced urinary potassium excretion
(Fig 2) with consequent increase in serum potassium.
With increased potassium retention, the excretion of
hydrogen is impaired leading to metabolic acidosis.
Hypertension tends to appear later in life, in men
aged 30-40 years and in women aged 40-50 years,5
but can also be absent in 20% of cases.3
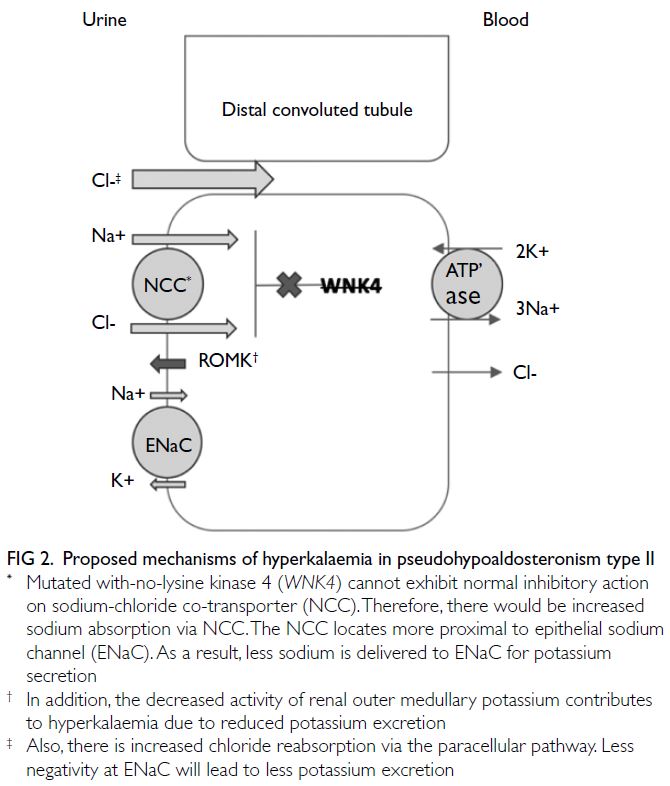
Figure 2. Proposed mechanisms of hyperkalaemia in pseudohypoaldosteronism type II
In this patient, pseudohypoaldosteronism
type I was initially suspected in view of the grossly
elevated renin and aldosterone levels. However,
the age of disease onset and normal sodium level
were atypical. The elevated renin and aldosterone
levels in our patient could be explained by the use of
furosemide that could have resulted in hypovolaemia
and thus activation of the renin-angiotensin-aldosterone-system. This made the interpretation of
the biochemical picture challenging. In this regard,
the use of genetic testing demonstrated superiority
in guiding the diagnosis of these chronic conditions
in which a classic clinical phenotype had already
been modified by existing treatments.
Our case also highlights the importance of
cascade family screening. The three family members
were identified to have potentially life-threatening
hyperkalaemia and were treated promptly based
on their genetic mutation. Affected patients will continue regular follow-up with serum electrolyte
and blood pressure monitoring. With specific
treatment, the disease can be well-controlled.
This case illustrates the importance of precision
medicine, as well as its benefits of personalised and
targeted interventions.
Author contributions
Concept or design: HY Lam, ALT Ma.
Acquisition of data: YH Chan, ALT Ma.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: HY Lam, EYH Chan.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: YH Chan, ALT Ma.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: HY Lam, EYH Chan.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
The genetic investigation was supported by the Health and Medical Research Fund (HMRF) by the Hong Kong Food and Health Bureau (Project Ref No: 06172806) and The Society for
the Relief of Disabled Children.
Ethics approval
The patient was treated in accordance with the tenets of the Declaration of Helsinki. The father of the patient consented verbally to publication of this case report.
References
1. Chung CC, Leung GK, Mak CC, et al. Rapid whole-exome
sequencing facilitates precision medicine in paediatric rare
disease patients and reduces healthcare costs. Lancet Reg
Health West Pac 2020;1:100001. Crossref
2. Paver WK, Pauline GJ. Hypertension and hyperpotassaemia
without renal disease in a young male. Med J Aust
1964;2:305-6. Crossref
3. Healy JK. Pseudohypoaldosteronism type II: history,
arguments, answers, and still some questions. Hypertension
2014;63:648-54. Crossref
4. Gordon RD. Syndrome of hypertension and hyperkalemia
with normal glomerular filtration rate. Hypertension
1986;8:93-102. Crossref
5. Farfel A, Mayan H, Melnikov S, Holtzman EJ, Pinhas-Hamiel O, Farfel Z. Effect of age and affection status on
blood pressure, serum potassium and stature in familial
hyperkalaemia and hypertension. Nephrol Dial Transplant
2011;26:1547-53. Crossref