© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Acquired C1-inhibitor deficiency in chronic
lymphocytic leukaemia: a case report
Andy SO Tang, MD, MRCP (UK)1; QY Wong, MB, BS2; ST Yeo, BPharm (Hon)2; Jenny TH Lee, MB, BS3; TS Leong, MB, BS, MRCP (UK)4; LP Chew, MD, FRCP (UK)4
1 Haematology Unit, Department of Internal Medicine, Miri Hospital, Ministry of Health, Sarawak, Malaysia
2 Department of Internal Medicine, Miri Hospital, Ministry of Health, Sarawak, Malaysia
3 Department of Pathology, Sarawak General Hospital, Ministry of Health, Sarawak, Malaysia
4 Haematology Unit, Department of Internal Medicine, Sarawak General Hospital, Ministry of Health, Sarawak, Malaysia
Corresponding author: Dr Andy SO Tang (andytangsingong@gmail.com)
 Full paper in PDF
Full paper in PDF
Case report
Acquired C1-inhibitor deficiency is a rare disorder
with varying manifestations and an estimated
prevalence of 1 per 100 000 to 500 000 population.1
We present a case of acquired angio-oedema that
manifested after a diagnosis of chronic lymphocytic
leukaemia (CLL).
In June 2017, a 60-year-old man who
was previously healthy was found to have
lymphocytosis on routine health screening. Physical
examination showed multiple lymph nodes with
no hepatosplenomegaly. A lymph node biopsy was
consistent with small lymphocytic lymphoma. Bone
marrow examination revealed small mature-looking
lymphocytes with a background of smudge cells.
The leukaemia cells co-expressed the CD5 antigen
and B-cell surface antigens, with restricted kappa
immunoglobulin light chain expression. Fluorescence
in situ hybridisation analysis demonstrated a
homozygous 13q14.2 gene deletion. Serum kappa and lambda free light chain (FLC) results were
99.50 mg/L (reference 6.70-22.40 mg/L) and
12.50 mg/L (reference 8.30-27.00 mg/L),
respectively, with a kappa to lambda ratio of
7.96. Beta-2 microglobulin level was 4.64 mg/L
(reference 1.09-2.53 mg/L). The patient screened
seronegative for human immunodeficiency virus,
syphilis, and hepatitis B and C. Overall findings
were consistent with a diagnosis of CLL Rai stage II
(Binet stage B), with Eastern Cooperative Oncology
Group performance status zero. A ‘wait and watch’
approach was adopted initially until mid-2018
when the patient started to report fatigue and
absolute lymphocyte count doubled in less than
6 months. Owing to limited resources, the patient
was given weekly intravenous rituximab (500 mg)
but developed herpes zoster infection after two
cycles. Treatment was changed to chlorambucil
(8 mg once daily) and prednisolone (20 mg once
daily) for 10 days per cycle, repeating every 28 days,
with acyclovir prophylaxis.
At 1 year after this treatment started, the
patient presented with an acute event of angio-oedema
that began as a tingling around his face and
mouth similar to a bee sting and swelling of his lips
and tongue (Fig 1), sparing the larynx, abdomen,
extremities, and genital involvement. There were
no identifiable inciting episodes. He denied intake
of angiotensin-converting enzyme inhibitors,
traditional medications, or over-the-counter drugs.
He had no history of atopy or angio-oedema.
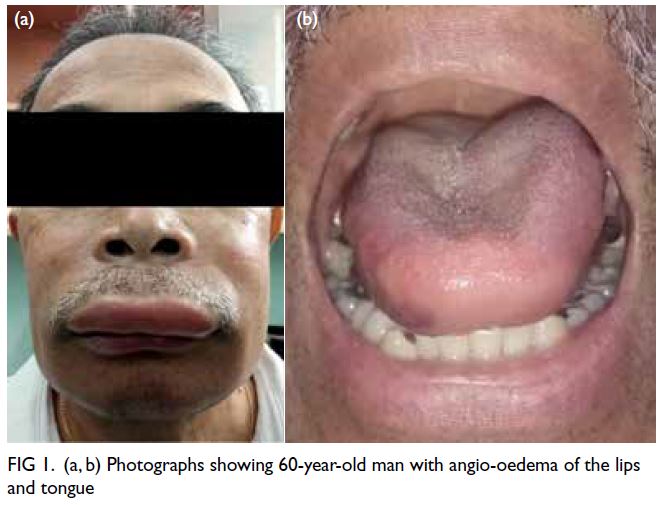
Figure 1. (a, b) Photographs showing 60-year-old man with angio-oedema of the lips and tongue
The patient remained well until early 2020
when he presented with similar episodes of angio-oedema
that recurred on three occasions. A complete
workup revealed a C1 esterase inhibitor antigen level
of 4 mg/dL (reference 19-37 mg/dL) with function
being 9% (reference <66%). Complement C1q level
measured using radial immunodiffusion assay was
undetectable with low complement 3 and 4 [0.29 g/L
(reference value 0.9-1.8 g/L), <0.02 g/L (reference
value 0.15-0.45 g/L), respectively]. Owing to
restricted resources, anti-C1 inhibitor antibodies and
monoclonal component isotypes were unavailable. Autoimmune screening (antinuclear antibody and
rheumatoid factor) and serum cryoglobulin were
negative. Skin biopsy showed features of angio-oedema
without atypical lymphocytes (Fig 2).
During the fourth acute attack of angio-oedema, the
patient was given tranexamic acid and danazol and
symptoms completely resolved within 24 hours.
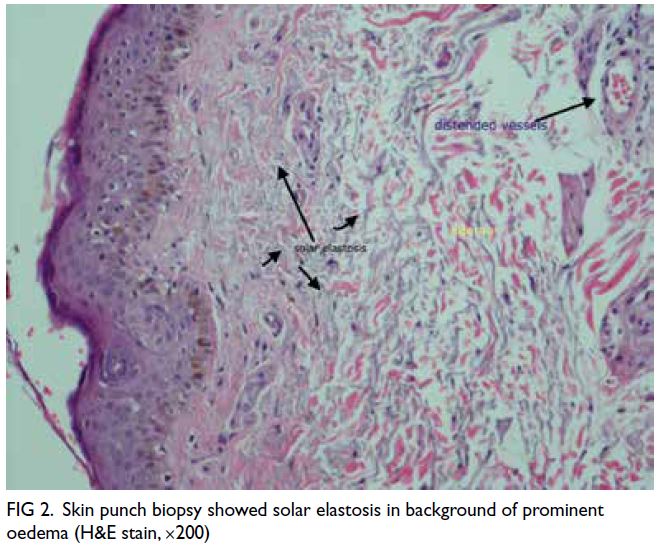
Figure 2. Skin punch biopsy showed solar elastosis in background of prominent oedema (H&E stain, ×200)
The overall clinical manifestations and
laboratory findings were consistent with the diagnosis
of acquired angio-oedema due to C1 deficiency
in association with CLL. We decided to initiate
chemotherapy—rituximab, cyclophosphamide,
vincristine, and prednisolone. After two cycles, the
patient developed urticaria vasculitis over his limbs
that responded to cetirizine and resolved completely
after completing six cycles of chemotherapy. C1
esterase inhibitor functional assay normalised. He
remained free of any recurrent angio-oedema and
urticaria during the 1-year follow-up period. He was
able to resume work and could live independently.
Discussion
Angio-oedema can be hereditary or acquired.
The latter often presents after the fifth decade of
life with no family history.1 Symptom onset and
absence of family history led to a diagnosis of
acquired angio-oedema in our patient due to C1
esterase inhibitor deficiency. The median time to
diagnosis is reported to vary between 10 months
and 10 years.2 Our patient was diagnosed 9 months
after symptom onset and during the third episode
of angio-oedema. To the best of our knowledge, only about 20 cases of CLL have been reported to
be associated with angio-oedema.2 Many diseases
are reported to be associated with acquired angio-oedema,
with lymphoproliferative disorders and
B-cell malignancies the most common.3
In acquired angio-oedema, C1 esterase
inhibitor protein level is decreased due to excessive
consumption, or may be dysfunctional due to the
presence of antibodies against C1 inhibitor, leading
to defective inhibition of the complements and kinin
system. This results in exaggerated response in the
classic complement pathway and production of
bradykinin leading to angio-oedema, in which C1q
and C4 level is low, as in our case.2 Our patient also
developed urticaria vasculitis. This was likely due to
C1q depletion and low complement, as previously
reported.2 Urticaria vasculitis, which likely involves
tumour-associated immune complexes, has been
described as a rare association with haematological
malignancy.4 The achievement of disease remission
led to resolution of both angio-oedema and urticarial
vasculitis in our case.
Our patient demonstrated a kappa-restricted
serum FLC with the summated FLC being 112 mg/L,
confirming the monoclonal nature of serum FLC.
Studies have shown that serum FLC may be elevated
in almost 50% of patients with CLL. This finding is
an adverse prognostic factor for time to treatment
and overall survival.5
The mainstay of treatment for acute acquired
angio-oedema includes C1 esterase inhibitor
concentrate from human plasma or recombinant
C1 inhibitor concentrate.3 Nonetheless these
products are not widely available in most healthcare
facilities, including our centre where fresh frozen
plasma is a reasonable alternative, although not
without potential risks. Long-term prophylactic
treatment with antifibrinolytic agents and danazol
has been shown to be equally efficacious,2 with
a recommendation to avoid the former in the
presence of thromboembolic risk factors.2 It is vital
to treat the underlying disease to prevent acute
attacks, regardless of the treatment options.2 Our
patient has had no further attacks of angio-oedema
after receiving rituximab, cyclophosphamide,
vincristine, and prednisolone chemotherapy. This
is in concordance with the literature that reports
resolution of angio-oedema after treatment of the
underlying malignancy.4
Our case highlights the challenge posed in
managing a patient with CLL and atypical features.
Clinical suspicion should be heightened in patients
who present with recurrent episodes of angio-oedema.
Treating the underlying malignancy often
leads to complete resolution of symptoms.
Author contributions
Concept or design: ASO Tang, TS Leong, LP Chew.
Acquisition of data: ASO Tang, JTH Lee, QY Wong, ST Yeo.
Analysis or interpretation of data: JTH Lee, QY Wong, ST Yeo.
Drafting of the manuscript: ASO Tang, TS Leong, LP Chew.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: ASO Tang, JTH Lee, QY Wong, ST Yeo.
Analysis or interpretation of data: JTH Lee, QY Wong, ST Yeo.
Drafting of the manuscript: ASO Tang, TS Leong, LP Chew.
Critical revision of the manuscript for important intellectual content: All authors.
Conflicts of interest
The authors declare that they have no conflict of interest or financial disclosure.
Acknowledgement
The authors would like to thank the Director General of Health Malaysia and Clinical Research Centre (CRC) Miri
Hospital for permission to publish this paper.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
This article does contain studies with human participants and was registered via National Medical Research Register Malaysia with a Research ID of NMRR-20-1666-56020.
The patient(s) provided written informed consent for all
treatments and procedures and consent for publication.
References
1. Pappalardo E, Cicardi M, Duponchel C, et al. Frequent
de novo mutations and exon deletions in the C1inhibitor
gene of patients with angioedema. J Allergy Clin Immunol
2000;106:1147-54. Crossref
2. Gobert D, Paule R, Ponard D, et al. A nationwide study of
acquired C1-inhibitor deficiency in France: characteristics
and treatment responses in 92 patients. Medicine
(Baltimore) 2016;95:e4363. Crossref
3. Cicardi M, Zingale LC, Pappalardo E, Folcioni A, Agostoni A.
Autoantibodies and lymphoproliferative diseases in
acquired C1-inhibitor deficiencies. Medicine (Baltimore)
2003;82:274-81. Crossref
4. Marder RJ, Rent R, Choi EY, Gewurz H. C1q deficiency
associated with urticarial-like lesions and cutaneous
vasculitis. Am J Med 1976;61:560-5. Crossref
5. Sarris K, Maltezas D, Koulieris E, et al. Prognostic
significance of serum free light chains in chronic
lymphocytic leukemia. Adv Hematol 2013;2013:359071. Crossref