© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Combined pulmonary fibrosis and emphysema: a commonly missed diagnosis
KO Cheung, MBBS, FRCR; CC Chan, FRCR, FHKAM (Radiology)
Department of Radiology, North District Hospital, Hong Kong
Corresponding author: Dr KO Cheung (ronald.mbbs@gmail.com)
 Full paper in PDF
Full paper in PDF
In September 2019, a 79-year-old man was referred
to the medical out-patient clinic for assessment of
chronic cough and exertional shortness of breath.
He was an ex-smoker for more than 10 years and
previously worked as a bird market hawker. He
stopped working 6 months previously because
of coughing. He had no history of chemical or
occupational dust exposure and took no drugs
associated with pulmonary fibrosis. He had a known
history of previous pulmonary tuberculosis. The
patient had no history of fever, rash, polyarthralgia
or uveitis. He had no family history of autoimmune
disease. Physical examination revealed no rash or
joint pain and no ulcers. He had full proximal and
distal muscle power and no features of autoimmune
disorder.
Immunological tests revealed rheumatoid
factor, <15.9 IU/mL (normal range <15.9IU/mL);
anti-proteinase 3, 5.5 RU/mL (normal range
<20 RU/mL); and anti-myeloperoxidase, 3.7 RU/mL
(normal range <20 RU/mL). Immunological tests
were positive for anti-neutrophil cytoplasmic
antibodies, but negative for anti-nuclear antibodies,
anti-ds DNA, and anti-extractable nuclear antigen.
Echocardiogram was not performed.
Lung function testing performed in September
2019 revealed a forced expiratory volume in 1
second (FEV1) of 2.23 L (98% predicted), forced
vital capacity (FVC) 3.01 L (95% predicted) and
FEV1/FVC ratio 74.1% (above predicted). Results of
lung function tests suggested that the shortness of
breath was likely due to a combination of restrictive
and obstructive lung defects (the former plays a
dominant role). The patient subsequently underwent
chest radiography and computed tomographic (CT)
imaging.
Chest radiograph in 2020 (Fig 1a) showed
coarsened bilateral lower lobe interstitial markings,
raising a concern for a superimposed parenchyma
process, such as pulmonary oedema or other
chronic process such as fibrosis. Reviewing patient’s
previous chest radiograph in 2009 (Fig 1b), there was
no bilateral lower lobe interstitial markings. This
demonstrated that bilateral lower lobe interstitial
markings in the 2020 chest radiograph were recent
onset.
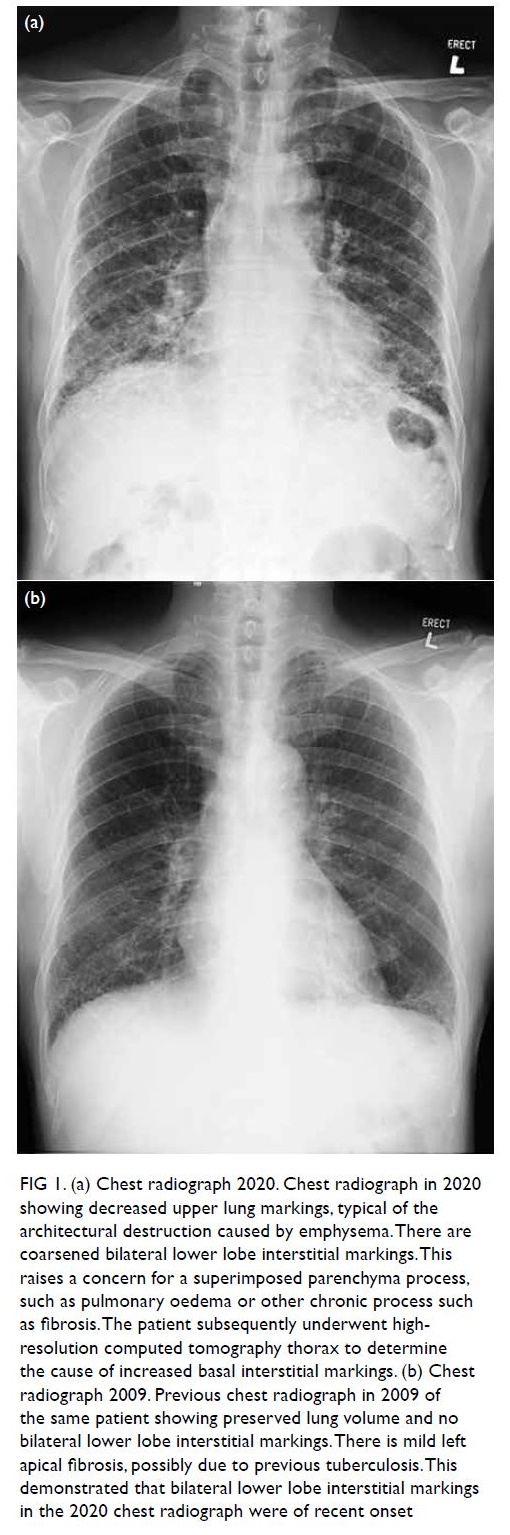
Figure 1. (a) Chest radiograph 2020. Chest radiograph in 2020 showing decreased upper lung markings, typical of the architectural destruction caused by emphysema. There are coarsened bilateral lower lobe interstitial markings. This raises a concern for a superimposed parenchyma process, such as pulmonary oedema or other chronic process such as fibrosis. The patient subsequently underwent highresolution computed tomography thorax to determine the cause of increased basal interstitial markings. (b) Chest radiograph 2009. Previous chest radiograph in 2009 of the same patient showing preserved lung volume and no bilateral lower lobe interstitial markings. There is mild left apical fibrosis, possibly due to previous tuberculosis. This demonstrated that bilateral lower lobe interstitial markings in the 2020 chest radiograph were of recent onset
Computed tomography thorax (Fig 2) in 2020
showed centrilobular and paraseptal emphysematous
change at bilateral upper zones. There was septal
thickening with reticulations, honeycomb formation and mild traction bronchiectasis at basal regions. On
the basis of these findings, a radiological diagnosis of
pulmonary fibrosis with emphysema was made.
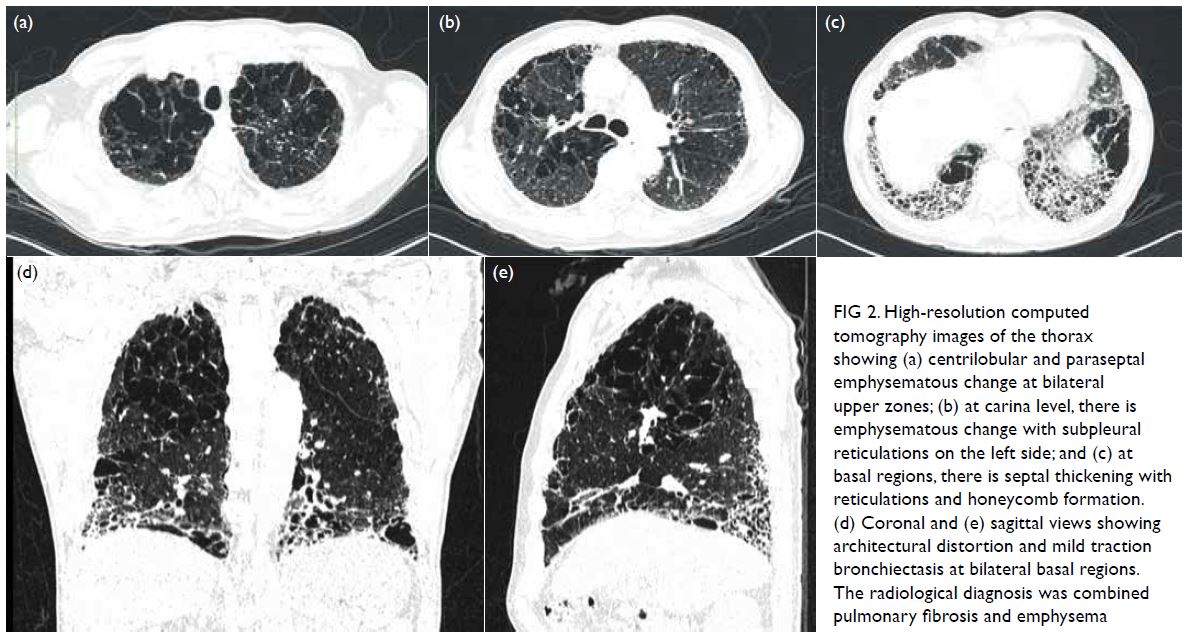
Figure 2. High-resolution computed tomography images of the thorax showing (a) centrilobular and paraseptal emphysematous change at bilateral upper zones; (b) at carina level, there is emphysematous change with subpleural reticulations on the left side; and (c) at basal regions, there is septal thickening with reticulations and honeycomb formation. (d) Coronal and (e) sagittal views showing architectural distortion and mild traction bronchiectasis at bilateral basal regions. The radiological diagnosis was combined pulmonary fibrosis and emphysema
Pulmonary function testing on 17 January
2020 revealed severely diminished diffusing capacity
for carbon monoxide (DLCO) of 35% (predicted:
19.1 mL/mmHg/min, best: 6.6 mL/mmHg/min)
and carbon monoxide diffusion coefficient of
41% (predicted: 4.29 mL/mHg/min/L, best:
1.74 mL/mHg/min/L). Results of testing
demonstrated no airflow obstruction or significant
post-bronchodilator response. The patient’s
DLCO and carbon monoxide diffusion coefficient
were low, indicating impaired diffusion due to
underlying pulmonary fibrosis. Based on his
DLCO <80% predicted and FEV1 >80% predicted,
cardiopulmonary exercise testing was proposed to
determine any need for lung resection.
Radiological and clinical
significance of combined pulmonary fibrosis and emphysema
Characteristic radiological findings of combined
pulmonary fibrosis and emphysema (CPFE)
syndrome include upper-lobe emphysema and
lower-lobe interstitial fibrotic changes. The
emphysema in CPFE includes bullous, paraseptal,
and centrilobular changes and is typically distributed
in the upper lobes. Fibrotic changes are not typical in
emphysema and should prompt further aetiological
investigation. Honeycombing refers to CT-detected
clustered thick-wall cystic air spaces (3 to 10 mm
in diameter, but occasionally as large as 25 mm)
that are usually subpleural, peripheral and basal in
distribution. Honeycombing indicates interstitial
fibrosis. In our patient, bilateral basal honeycombing
on CT confirmed end-stage fibrosis as the cause
of increased interstitial markings seen on chest
radiography.
The coexistence of pulmonary fibrosis
and emphysema was first noted in 1990 but was
not considered a distinct entity until further
characterisation 15 years later. There has been
increasing recognition that these two processes
may coexist in some patients, and this overlapping
disorder has often been termed combined
emphysema and fibrosis or CPFE. In general, patients
with CPFE have preserved FEV1 and FVC, but the diffusion capacity of the lung for carbon monoxide is
severely diminished.1
Typically, CPFE is more common in men,
current or former smokers.2 Some classic features of
CPFE include the following:
Management of combined
pulmonary fibrosis and emphysema
The mainstay of treatment for patients with
CPFE is supportive care. Smoking cessation is
definitely indicated for both components of CPFE.
Supplemental oxygen therapy may be beneficial,
also COPD treatments such as bronchodilators
and inhaled steroids. Case reports of patients with
CPFE reveal that lung volume reduction (LVR)
may be beneficial in cases of advanced emphysema,
even without plethysmographic evidence of severe
hyperinflation.5 Treatment with antifibrotic drugs,
such as pirfenidone and nintedanib, may be effective
in CPFE but further trials are awaited.2 There is
evidence that nintedanib can decrease the annual
rate of decline in FVC in patients with other (non-usual
interstitial pneumonia-like) fibrotic patterns
as well as those with IPF. Currently, to the best
of our knowledge these are not yet available for
CPFE.2 Further investigation is needed into future
use of antifibrotic drugs for CPFE. Ultimately, lung
transplantation is the only cure.
Author contributions
All authors contributed to the concept, acquisition and
interpretation of data, drafting of the manuscript, and
revision for important intellectual content. All authors had
full access to the data, contributed to the study, approved the
final version for publication, and take responsibility for its
accuracy and integrity.
Conflicts of interest
All authors declare no conflicts of interest related to the work in this manuscript.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
This study was conducted in accordance with the principles outlined in the Declaration of Helsinki. The patient provided
written informed consent for all treatments and procedures,
and verbal consent for the publication of this study.
References
1. Cottin V. Combined pulmonary fibrosis and emphysema: bad and ugly all the same? Eur Respir J 2017;50:1700846. Crossref
2. Hage R, Gautschi F, Steinack C, Schuurmans MM. Combined pulmonary fibrosis and emphysema (CPFE)
clinical features and management. Int J Chron Obstruct
Pulmon Dis 2021:16:167-77. Crossref
3. Kitaguchi Y, Fujimoto K, Hanaoka M, Kawakami S, Honda T, Kubo K. Clinical characteristics of combined
pulmonary fibrosis and emphysema. Respirology
2010;15:265-71. Crossref
4. Cottin V, Nunes H, Brillet PY, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised
entity. Eur Respir J 2005;26:586-93. Crossref
5. Straub G, Caviezel C, Frauenfelder T, Bloch KE, Franzen D.
Successful lung volume reduction surgery in combined
pulmonary emphysema and fibrosis without body-plethysmographic
hyperinflation—a case report. J Thorac
Dis 2018;10 (Suppl 23):S2830-4. Crossref