© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Isolated hereditary diffuse palmoplantar
keratoderma in Hong Kong Chinese patients: a case series
PT Yu, MB, BS, FHKAM (Paediatrics)1; Stephanie Ho, MB, ChB, FHKAM (Paediatrics)1; SC Ng, MB, BS, FHKAM (Medicine)2; FM Lo, MB, BS, FHKAM (Paediatrics)1; HM Luk, MB, BS, FHKAM (Paediatrics)1
1 Clinical Genetic Service, Department of Health, Hong Kong
2 Social Hygiene Clinic, Department of Health, Hong Kong
Corresponding author: Dr HM Luk (luksite@gmail.com)
 Full paper in PDF
Full paper in PDF
Case report
Patient 1
A 26-year-old man presented with a history of
hyperhidrosis of the palms and soles since age 6
months. He subsequently developed progressive
palmoplantar hyperkeratosis. He enjoyed
otherwise good past health. His parents were non-consanguineous
and there was no significant family
history. Physical examination revealed bilateral
symmetrical thick waxy hyperkeratosis of the palms
and soles with well-demarcated transgradient
margins and focal macerated erosions over the
palms. Knuckle pads were observed on the dorsum
of the proximal interphalangeal joints of the fingers
and toes (Fig 1). There were no associated ectodermal
manifestations, brachydactyly, nail abnormality
or deformity. There was no insolate lesion over
the elbows or knees. Medical exome sequencing
identified homozygous pathogenic variants
in NM_020427.2(SLURP1) c.147_150delCTGC,
p.Cys50*. The molecular diagnosis of Meleda disease
[MIM #248300] was confirmed.
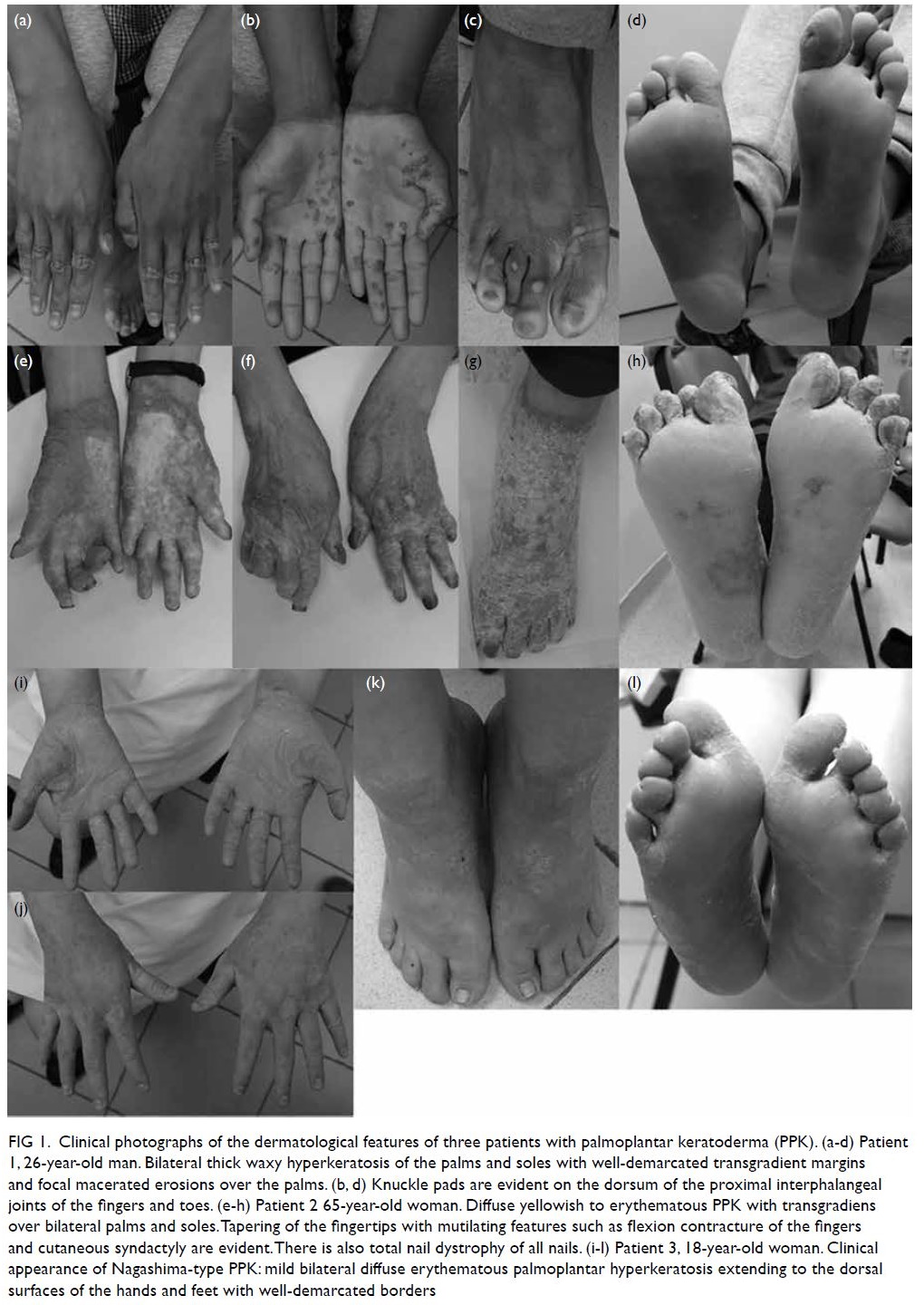
Figure 1. Clinical photographs of the dermatological features of three patients with palmoplantar keratoderma (PPK). (a-d) Patient 1, 26-year-old man. Bilateral thick waxy hyperkeratosis of the palms and soles with well-demarcated transgradient margins and focal macerated erosions over the palms. (b, d) Knuckle pads are evident on the dorsum of the proximal interphalangeal joints of the fingers and toes. (e-h) Patient 2 65-year-old woman. Diffuse yellowish to erythematous PPK with transgradiens over bilateral palms and soles. Tapering of the fingertips with mutilating features such as flexion contracture of the fingers and cutaneous syndactyly are evident. There is also total nail dystrophy of all nails. (i-l) Patient 3, 18-year-old woman. Clinical appearance of Nagashima-type PPK: mild bilateral diffuse erythematous palmoplantar hyperkeratosis extending to the dorsal surfaces of the hands and feet with well-demarcated borders
Patient 2
A 65-year-old woman presented with a history of
diffuse hyperkeratosis of bilateral palms and soles
since early infancy. She otherwise enjoyed good
past health. Her parents were first-degree cousins
and her younger brother had similar dermatological
features. Skin biopsy revealed non-epidermolytic
keratoderma. Physical examination showed
diffuse yellowish to erythematous palmoplantar
keratoderma (PPK) with transgradiens over bilateral
palms and soles and well-demarcated margins. The
fingers were mutilating with camptodactyly and
cutaneous syndactyly. There was total nail dystrophy
of all fingernails and toenails but no insolate lesions
over the elbows or knees (Fig 1). Whole exome
sequencing revealed homozygous likely pathogenic
variants in SLURP1 NM_020427.2(SLURP1)
c.256G>A, p.Gly86Arg. The molecular diagnosis of
Meleda disease [MIM #248300] was confirmed.
Patient 3
An 18-year-old woman presented with mild bilateral
diffuse erythematous palmoplantar keratosis
extending to the dorsal surfaces of the hands
and feet since early infancy (Fig 1). There was no
associated systemic involvement and no significant
family history. Skin biopsy (Fig 2) revealed marked
compact orthokeratosis, focal parakeratosis, mild
hypergranulosis, acanthosis and psoriasiform
epidermal hyperplasia, as well as mononuclear
inflammatory infiltrate in the dermis.

Figure 2. Histology of skin biopsy from Patient 3 showing (a) marked compact orthokeratosis (closed arrows), acanthosis and psoriasiform epidermal hyperplasia (open arrows), and focal parakeratosis (arrowheads) [haematoxylin and eosin, ×25], and (b) mild hypergranulosis (closed arrow) and mononuclear inflammatory infiltrate (arrowheads) in the dermis (haematoxylin and eosin, ×100)
Medical exome sequencing
identified biallelic pathogenic variants in
NM_001040147.2(SERPINB7):c.169-1G>A, p.? and
c.522dup, p.Val175Cysfs*46. The molecular diagnosis
of PPK, Nagashima type [MIM #615598] was
confirmed.
Discussion
Hereditary PPK is a heterogeneous group of disorders
characterised by marked hyperkeratosis of the palms
and soles due to a defect in cornification. It can occur
in isolation or in association with other ectodermal
defects or extracutaneous manifestations. Molecular
studies are sometimes crucial to reach an accurate
subtype classification as there are significant
overlapping clinical features and heterogeneity
among different types of hereditary PPK. Depending
on the morphology of the lesions, PPK is classified
into three major patterns: diffuse, focal (areata or
striata), and punctate. Although precise figures of
incidence are lacking, PPK is generally perceived to
be rare. In Asia, the most frequent type of PPK is
Nagashima PPK, with an estimated prevalence of
3.1 per 10 000 population.1 This article summarises
the clinical features and molecular findings in three
Chinese individuals with isolated diffuse hereditary
PPK recruited from a single centre and highlights
the significance of genetic testing in reaching an
accurate classification and diagnosis.
Meleda disease (also known as Mal de Meleda)
has an autosomal recessive pattern of inheritance and is characterised by an early onset of bilateral,
diffuse, well-demarcated thick and waxy PPK
in a glove and stocking pattern. It is rare, with a
prevalence of 1:100 000.2 Palmoplantar erythema can
be evident from early infancy and tends to progress
to thick hyperkeratosis as an affected individual
ages. Meleda disease is commonly associated with
hyperhidrosis and nail anomalies such as koilonychia,
onychogryphosis, and subungual hyperkeratosis.
Anomalies of the digits, including pseudoainhum,
contracture, tapering of digits, knuckle pads
and fifth-finger dysplasia have been reported.
Oral manifestations including lower-lip angular
cheilitis, high arch palate and perioral erythema
can be variably present. Skin biopsy is characterised
by histological findings of hyperkeratosis and
acanthosis in the epidermis without evidence of
epidermolysis.2 SLURP-1 was subsequently identified
to be responsible for Meleda disease in 2001 by Fischer et al.3 SLURP-1 is involved in regulation of
inflammation and keratinocyte apoptosis. Treatment
options include oral retinoids, topical keratolytics,
and surgical excision of hyperkeratosis followed by
placement of a full-thickness skin graft. Infection
is treated with antibacterials and antifungals, with
some centres advocating the use of prophylactic
topical antifungals due to a susceptibility to tinea
infection.
Nagashima-type PPK (NPPK) is probably the
most common form of PPK among patients of Asian
ethnicity. It was initially described by Nagashima4 in
1977. Biallelic mutations of SERPINB7 were found to
be causative of NPPK in 2013.5 The hyperkeratosis is
non-progressive after puberty and has milder clinical
features when compared with Meleda disease. It is
characterised by diffuse mild erythematous non-mutilating
palmoplantar hyperkeratosis that can
transform into a spongy white appearance after
immersion in water. The dorsal surface of the hands
and feet, ankles, Achilles tendon area, elbows and
knees can be involved. Palmoplantar hyperkeratosis
occurs in isolation without associated ectodermal
or extracutaneous manifestations. Histological
findings are unremarkable with orthohyperkeratosis,
acanthosis and mild perivascular inflammatory
infiltration of lymphocytes in the upper dermis.1 A
nonsense c.796C>T founder mutation in SERPINB7
has been reported to be prevalent in Chinese
patients with NPPK,1 but c.169-1G>A in Patient 3
is novel to the literature. No curative treatment is
available but topical application of gentamicin has
been investigated and appears promising.6
Conclusion
We have reported three cases of PPK that illustrate
the clinical phenotype and molecular findings of this
particular subgroup of genodermatoses. Molecular
investigations may be warranted for accurate
diagnosis of PPK and to determine its inheritance,
and may be beneficial in reproductive management
within the family.
Author contributions
All authors contributed equally to the concept or design of the study, acquisition of the data, analysis or interpretation of
the data, drafting of the manuscript, and critical revision of
the manuscript for important intellectual content. All authors
had full access to the data, contributed to the study, approved
the final version for publication, and take responsibility for its
accuracy and integrity.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Acknowledgement
The authors thank Dr WC Siu for preparing the histology photographs.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patients were treated in accordance with the Declaration of Helsinki. The patients provided written informed consent
for all treatments and procedures, and for publication of this
report.
References
1. Yin J, Xu G, Wang H, et al. New and recurrent SERPINB7 mutations in seven Chinese patients with Nagashima-type palmoplantar keratosis. J Invest Dermatol 2014;134:2269-72. Crossref
2. Perez C, Khachemoune A. Mal de Meleda: a focused review. Am J Clin Dermatol 2016;17:63-70. Crossref
3. Fischer J, Bouadjar B, Heilig R, et al. Mutations in the gene encoding SLURP-1 in Mal de Meleda. Hum Mol Genet 2001;10;875-80. Crossref
4. Nagashima M. Palmoplantar keratosis. In: Miura O, Ochiai K, editors. Handbook of Human Genetics [in Japanese].
Vol 9. Tokyo: Igaku Shoin; 1977: 23-7.
5. Kubo A, Shiohama A, Sasaki T, et al. Mutations in SERPINB7, encoding a member of the serine protease
inhibitor superfamily, cause Nagashima-type palmoplantar
keratosis. Am J Hum Genet 2013;93:945-56. Crossref
6. Ohguchi Y, Nomura T, Suzuki S, et al. Gentamicin-induced readthrough and nonsense-mediated mRNA decay of SERPINB7 nonsense mutant transcripts. J Invest Dermatol 2018;138:836-43. Crossref