© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Next-generation sequencing panel for diagnosis and
management of chronic neutrophilic leukaemia: a case report
KY Mak, MB, ChB1; CH Au, PhD2;
TL Chan, PhD2; Edmond SK Ma, MD, FHKAM (Pathology)2;
Eudora YD Chow, MB, BS, FHKAM (Pathology)1; SY Lin, MB, BS,
FHKAM (Medicine)3; William WL Choi, MB, BS, FHKAM (Pathology)1
1 Department of Pathology, United
Christian Hospital, Kwun Tong, Hong Kong
2 Department of Pathology, Hong Kong
Sanatorium & Hospital, Happy Valley, Hong Kong
3 Department of Medicine and Geriatrics,
United Christian Hospital, Kwun Tong, Hong Kong
Corresponding author: Dr William WL Choi (wlchoi@hotmail.com)
 Full
paper in PDF
Full
paper in PDF
Case report
An 80-year-old woman presented to our hospital with
mild headache in November 2015. She had a history of hypertension,
diabetes mellitus, and hyperlipidaemia. She was febrile (38.6°C) but did
not appear septic. Abdominal examination revealed mild splenomegaly but no
hepatomegaly and there were no focal neurological signs or suggestions of
other organ involvement. A full blood count showed leukocytes 124.0 ×
109/L (neutrophils 120.3 × 109/L, lymphocytes 2.5 × 109/L,
monocytes 1.2 × 109/L); haemoglobin 7.8 g/dL, mean corpuscular volume 89.1
fL; and platelets 384 × 109/L. The blood film showed marked neutrophilia,
occasional myelocytes, and absolute basophilia but no blasts. The
neutrophils showed toxic granules and were not dysplastic (Fig
1). Plain radiographs of the chest, kidney, ureter, and urinary
bladder did not reveal any abnormalities. Bacterial cultures of the blood
and urine did not reveal any septic foci. Because of the marked
neutrophilia, the patient was initially treated for bacterial sepsis with
empirical intravenous amoxicillin with clavulanic acid. Subsequent
ultrasonography of the abdomen confirmed the splenomegaly (15.1 cm) but no
other space-occupying lesions. The low-grade fever soon subsided after
admission and she remained afebrile and non-septic, although the marked
neutrophilia persisted.
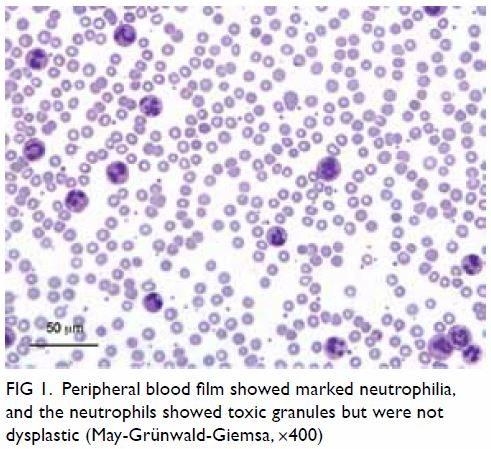
Figure 1. Peripheral blood film showed marked neutrophilia, and the neutrophils showed toxic granules but were not dysplastic (May-Grünwald-Giemsa, ×400)
A bone marrow biopsy revealed marked
hypercellularity, primarily due to markedly increased granulopoiesis with
left-shift in maturation but no increase in blasts. Erythropoiesis was
active and normoblastic. Megakaryocytes were moderately increased with
some being large and hyperlobulated. No overt dysplasia was seen.
Cytogenetic karyotyping showed a normal karyotype. Reverse transcription
polymerase chain reaction (PCR) for BCR-ABL1 and allele-specific
PCR for Janus kinase 2 (JAK2) V617F mutation analysis were
negative. In view of the clinical and laboratory picture of a possible
myeloproliferative neoplasm, and the lack of a clonal marker detected by
the molecular tests, we sought to utilise a next-generation sequencing
(NGS) panel (TruSight Myeloid Sequencing Panel; Illumina, San Diego [CA],
United States) to look for possible mutations in selected exons of 54
different genes commonly implicated in myeloid neoplasms, in accordance
with a previously published protocol.1
Deep sequencing by this panel returned three pathogenic mutations:
colony-stimulating factor 3 receptor (CSF3R) c.1853C>T;
p.Thr618Ile or T618I at variant allele frequency (VAF) of 43.9% (Fig
2a), serine/arginine-rich splicing factor 2 (SRSF2)
c.284C>T; p.Pro95Leu (NM_003016.4) or P95L at VAF of 49.6%, and
additional sex combs like 1 (ASXL1) truncating mutation c.1934dupG;
p.Gly646Trpfs*12 (NM_015338.5) at VAF of 34.1%. The CSF3R T618I mutation
was further confirmed by Sanger sequencing (Fig 2b), thus prompting the diagnosis of chronic
neutrophilic leukaemia (CNL). Although the CSF3R mutation rendered
the disease amenable to ruxolitinib, the patient deteriorated rapidly and
died of sudden severe gastrointestinal bleeding 12 days after admission
and before specific therapy could be contemplated.
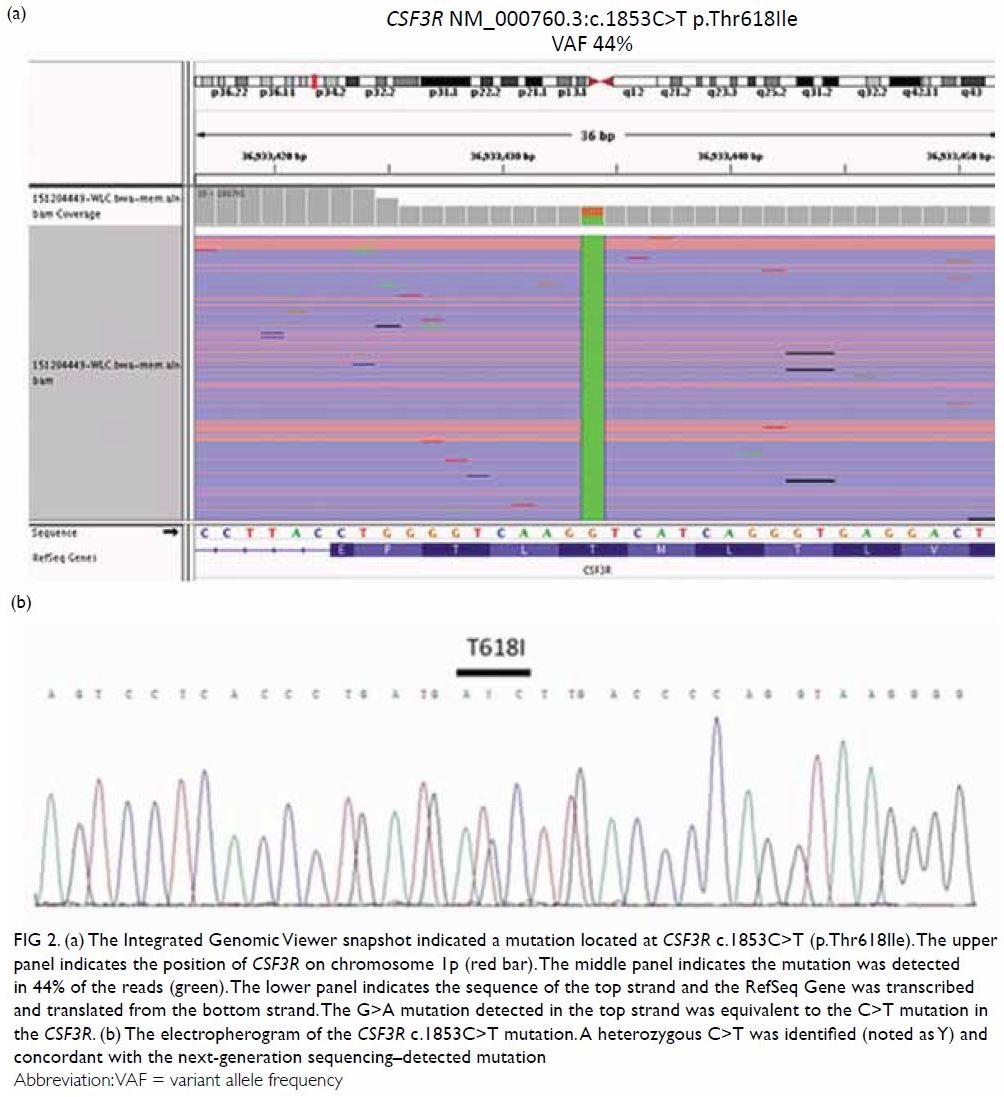
Figure 2. (a) The Integrated Genomic Viewer snapshot indicated a mutation located at CSF3R c.1853C>T (p.Thr618Ile). The upper panel indicates the position of CSF3R on chromosome 1p (red bar). The middle panel indicates the mutation was detected in 44% of the reads (green). The lower panel indicates the sequence of the top strand and the RefSeq Gene was transcribed and translated from the bottom strand. The G>A mutation detected in the top strand was equivalent to the C>T mutation in the CSF3R. (b) The electropherogram of the CSF3R c.1853C>T mutation. A heterozygous C>T was identified (noted as Y) and concordant with the next-generation sequencing–detected mutation
Discussion
Several clinical features in this patient hinted at
differential diagnoses other than bacterial infection or acute
inflammation. Apart from fever, there were no other clinical features
related to the presenting symptom of headache. Splenomegaly, a common
feature of myeloproliferative neoplasms, was present. The absolute
neutrophil count was very high, yet no clinical features of sepsis were
found on physical examination or from investigations.
Since 2013, understanding of the molecular genetics
of CNL has been dramatically changed by the discovery of CSF3R
mutations in around 80% of cases.2
The CSF3R encodes a transmembrane receptor for granulocyte
colony-stimulating factor 3, and plays a crucial role in the
differentiation and maturation of neutrophils.2
The CNL-associated mutations in CSF3R activate the receptor and
promote the proliferation and differentiation of neutrophils, leading to
the marked neutrophilia that characterises the CNL disease phenotype.3 There are two major types of CSF3R mutations in
CNL. The first encompasses point mutations in the extracellular or
transmembrane domains, of which the T618I mutation is the most common and
comprises the majority of mutations in CNL. The second type of CSF3R
mutation comprises nonsense or frameshift mutations leading to a premature
stop codon and truncation of the cytoplasmic domain of the receptor.2
Mutations of other genes have also been reported in
CNL. These can be grouped as SET binding protein 1 (SETBP1)
mutations, spliceosome mutations (eg, SRSF2), epigenetic modifier
mutations (eg, ASXL1), and signalling mutations (eg, JAK2).3 One previous study found that SRSF2
mutations occurred in three of 14 cases of CNL (21%). The SRSF2
mutations were previously associated with a worse prognosis in chronic
myelomonocytic leukaemia, but its effects on CNL are unclear.3 A significant proportion of CNL patients have been
shown to harbour ASXL1 (30%-60%). Similar to mutations in other
myeloid malignancies, ASXL1 mutations in CNL have been shown to
confer a poor prognosis.3
Although these other mutations have not been
incorporated into the World Health Organization 2016 diagnostic criteria
for CNL, these data suggest that some may show prognostic value.
Additionally, different CSF3R mutations may allow different
therapeutic approaches (see below). Because Sanger sequencing of an
increasing number of genes leads to substantial increases in the required
time, resources, and necessary amount of DNA, we sought to explore an NGS
panel to interrogate these genes simultaneously more efficiently and
cost-effectively. Of note, the panel includes genes that are important for
the diagnosis of myeloproliferative neoplasm (JAK2, calreticulin [CALR]
and myeloproliferative leukaemia protein [MPL]), plus genes that
are frequently reported in CNL (CSF3R, SETBP1, SRSF2,
ASXL1). Therefore, compared with Sanger sequencing, NGS panels are
a more efficient and powerful means to enable comprehensive genomic
profiling of individual CNL cases, utilising a smaller amount of DNA.
With recent discoveries in the molecular
pathogenesis of CNL, new therapeutic approaches that target the CSF3R
signalling pathway–related SRC family and JAK-kinase pathways have
emerged. The SRC signalling pathway is activated by truncation mutations
of CSF3R leading to sensitivity to dasatinib, while the JAK-STAT
pathway is activated by membrane proximal mutations of CSF3R
leading to sensitivity to ruxolitinib.2
Although there are few reported cases of these agents,2 4 they
represent significant breakthroughs in the management of CNL. This case of
a rare myeloproliferative neoplasm demonstrates how advances in
understanding of the molecular pathogenesis of a disease open up new
routes for the development of effective novel therapeutic strategies.
Author contributions
All authors had full access to the data,
contributed to the study, approved the final version for publication, and
take responsibility for its accuracy and integrity.
Concept and design of study: WWL Choi.
Acquisition of data: KY Mak, CH Au, TL Chan, ESK Ma, SY Lin, WWL Choi.
Analysis or interpretation of data: KY Mak, CH Au, TL Chan, ESK Ma, WWL Choi.
Drafting of the manuscript: KY Mak, WWL Choi.
Critical revision for important intellectual content: CH Au, TL Chan, ESK Ma, EYD Chow, SY Lin.
Acquisition of data: KY Mak, CH Au, TL Chan, ESK Ma, SY Lin, WWL Choi.
Analysis or interpretation of data: KY Mak, CH Au, TL Chan, ESK Ma, WWL Choi.
Drafting of the manuscript: KY Mak, WWL Choi.
Critical revision for important intellectual content: CH Au, TL Chan, ESK Ma, EYD Chow, SY Lin.
Conflicts of interest
All authors have no conflicts of interest to
declare.
Funding/support
This research received no specific grant from any
funding agency in the public, commercial, or not-for-profit sectors.
References
1. Au CH, Wa A, Ho DN, Chan TL, Ma ES.
Clinical evaluation of panel testing by next-generation sequencing (NGS)
for gene mutations in myeloid neoplasms. Diagn Pathol 2016;11:11. Crossref
2. Maxson JE, Gotlib J, Pollyea DA, et al.
Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical
CML. N Engl J Med 2013;368:1781-90. Crossref
3. Maxson JE, Tyner JW. Genomics of chronic
neutrophilic leukemia. Blood 2017;129:715-22. Crossref
4. Stahl M, Xu ML, Steensma DP, Rampal R,
Much M, Zeidan AM. Clinical response to ruxolitinib in CSF3R
T618-mutated chronic neutrophilic leukemia. Ann Hematol 2016;95:1197-200.
Crossref