Hong
Kong Med J 2019 Feb;25(1):21–9 | Epub 23 Jan 2019
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Sudden arrhythmia death syndrome in young victims: a
five-year retrospective review and two-year prospective molecular autopsy
study by next-generation sequencing and clinical evaluation of their
first-degree relatives
Chloe M Mak, MD, FHKAM (Pathology)1#; NS
Mok, FHKAM (Medicine), FRCP (Edin)2#; HC Shum, MRCP (UK)3;
WK Siu, PhD, FHKAM (Pathology)1; YK Chong, FHKCPath, FHKAM
(Pathology)1; Hencher HC Lee, FRCPath, FHKAM (Pathology)1; NC
Fong, FHKAM (Paediatrics)4; SF Tong, MSc1; KW Lee,
BNurs2; CK Ching, FRCPA, FHKAM (Pathology)1; Sammy
PL Chen, FRCPA, FHKAM (Pathology)1; WL Cheung, MB, BS1;
CB Tso, FHKCPath3; WM Poon, FHKCPath, FHKAM (Pathology)3;
CL Lau, FHKAM (Medicine)3; YK Lo, FHKAM (Medicine)3;
PT Tsui, FHKAM (Medicine)2; SF Shum, FHKCPath, FHKAM
(Pathology)3; KC Lee, MB, ChB, FHKAM (Pathology)1
1 Department of Pathology, Princess
Margaret Hospital, Kwai Chung, Hong Kong
2 Department of Medicine and Geriatrics,
Princess Margaret Hospital, Kwai Chung, Hong Kong
3 Forensic Pathology Service, Department
of Health, Hong Kong
4 Department of Paediatrics,
Princess Margaret Hospital, Kwai Chung, Hong Kong
# The first two authors contributed
equally to the work.
Corresponding author: Dr NS Mok (mokns@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Objective: Sudden arrhythmia
death syndrome (SADS) accounts for about 30% of causes of sudden cardiac
death (SCD) in young people. In Hong Kong, there are scarce data on SADS
and a lack of experience in molecular autopsy. We aimed to investigate
the value of molecular autopsy techniques for detecting SADS in an East
Asian population.
Methods: This was a two-part
study. First, we conducted a retrospective 5-year review of autopsies
performed in public mortuaries on young SCD victims. Second, we
conducted a prospective 2-year study combining conventional autopsy
investigations, molecular autopsy, and cardiac evaluation of the
first-degree relatives of SCD victims. A panel of 35 genes implicated in
SADS was analysed by next-generation sequencing.
Results: There were 289 SCD
victims included in the 5-year review. Coronary artery disease was the
major cause of death (35%); 40% were structural heart diseases and 25%
were unexplained. These unexplained cases could include SADS-related
conditions. In the 2-year prospective study, 21 SCD victims were
examined: 10% had arrhythmogenic right ventricular cardiomyopathy, 5%
had hypertrophic cardiomyopathy, and 85% had negative autopsy. Genetic
analysis showed 29% with positive heterozygous genetic variants; six
variants were novel. One third of victims had history of syncope, and
14% had family history of SCD. More than half of the 11 first-degree
relatives who underwent genetic testing carried related genetic
variants, and 10% had SADS-related clinical features.
Conclusion: This pilot
feasibility study shows the value of incorporating cardiac evaluation of
surviving relatives and next-generation sequencing molecular autopsy
into conventional forensic investigations in diagnosing young SCD
victims in East Asian populations. The interpretation of genetic
variants in the context of SCD is complicated and we recommend its
analysis and reporting by qualified pathologists.
New knowledge added by this study
- This study provides important data on the prevalence and types of sudden arrhythmia death syndrome (SADS) among young victims of sudden cardiac death in an East Asian population.
- This is the first local feasibility study on the service model incorporating cardiac evaluation of surviving relatives and molecular autopsy by next-generation sequencing into the conventional forensic investigations.
- Genomic testing should be conducted on patients with cardiomyopathies and channelopathies.
- Clinical assessment should be provided for at-risk family members irrespective of genetic findings.
- Molecular autopsy together with conventional autopsy conducted by qualified pathologists should be applied to victims of SADS, sudden unexpected death in epilepsy, or sudden infant death syndrome.
Introduction
Sudden death is defined as death occurring within 1
hour of the onset of symptoms or within 24 hours of the victim being seen
alive.1 Sudden death due to an
underlying heart disease is known as sudden cardiac death (SCD). The
worldwide annual incidence of SCD is about 4 to 5 million cases per year.2 A tragic and devastating
complication of a number of heart diseases, SCD is most often unexpected
and has major implications for the surviving family and the community. The
majority of SCD cases in middle-aged and older individuals are caused by
coronary artery disease; however, SCD is rare in young people and the
causes are more diversified.3 4 Autopsy studies have shown no structural heart disease
was found in up to 30% of young SCD victims.5
6 7
8
Molecular autopsy was first described by Ackerman
et al9 in 1999, to determine the
cause of death in uncertain cases after conventional autopsy by genetic
analysis. Post-mortem genetic studies have shown that SCD in these victims
can be caused by fatal arrhythmias secondary to a group of inheritable
cardiac electrical disorders collectively known as sudden arrhythmia death
syndrome (SADS).10 11 12 13 14 These
include Brugada syndrome (BrS), long QT syndrome (LQTS), short QT
syndrome, catecholaminergic polymorphic ventricular tachycardia (CPVT),
arrhythmogenic right ventricular cardiomyopathy (ARVC), hypertrophic
cardiomyopathy (HCM), and other cardiomyopathies.
Because SADS-related conditions are genetic
diseases, there are two different approaches to identify SADS among young
sudden unexplained death (SUD) victims. The first approach involves
detailed clinical and targeted genetic examination of the surviving
relatives of SCD victims. Studies using this approach suggest that SADS
may account for approximately 40% of autopsy-negative sudden death in
young people.10 15 However, this approach may not be able to identify
subjects with concealed form of SADS due to incomplete penetrance and
variable expressivity of the pathological mutations. The second approach
is to perform molecular autopsy on SCD victims, which involves post-mortem
genetic testing for SADS. A landmark study on molecular autopsy by the
Mayo Clinic showed over one third of SCD cases hosted a presumably
pathogenic mutations of cardiac ion channel diseases.8 16 Thus a
combined approach using both cardiac evaluation of surviving relatives and
molecular autopsy on SUD victims should give a higher yield on elucidating
the underlying causes of SUD.
In Hong Kong, there are scarce data on the
prevalence and types of SADS underlying SCD or SUD in young people. The
present study aimed to investigate the prevalence and types of SADS in an
East Asian population, and to perform a pilot study on incorporating
cardiac evaluation of surviving relatives and molecular autopsy by
next-generation sequencing (NGS) into conventional forensic
investigations.
Methods
In the present study, first, we carried out a
5-year retrospective review of the records of all autopsies for young
sudden death victims performed in public mortuaries in Hong Kong. Second,
we conducted a 2-year study to determine the prevalence and types of SADS
as the underlying causes of SCD among local young victims through
conventional and molecular autopsy, and evaluated their first-degree
relatives.
Five-year retrospective review of autopsy records
The Forensic Pathology Service, Department of
Health, provides all public autopsy services for over 7 million people in
Hong Kong. We performed this retrospective review of the records of all
autopsies in public mortuaries for young sudden death victims (aged 5-40
years) between 1 January 2008 and 31 December 2012. Sudden death was
defined as death occurring within 1 hour of the onset of symptoms or
within 24 hours of the victim being seen alive.1
Sudden death victims were recruited into the study for a detailed review
of their autopsy records. Data including age, height, weight, sex,
circumstances of death, clinical history of cardiac disease, and
pathologic findings at autopsy were collected and analysed. Sudden death
victims whose deaths were caused by trauma, accidents, drowning, and drug
toxicity, and those whose autopsy records were either incomplete or not
accessible for retrospective review were excluded.
Two-year prospective study by conventional and
molecular autopsy
In this prospective study, young SCD victims (aged
5-40 years) were identified and recruited into the study by forensic
pathologists after a finding of either an inheritable arrhythmogenic
cardiomyopathy or no anatomical cause of death (including other structural
heart disease) on autopsy, and a negative toxicology screening. Clinical
history, including personal history of arrhythmic events and history
surrounding the sudden death event of the SCD victim was collected during
identification interviews with next-of-kin as far as possible by forensic
pathologists. DNA-friendly blood samples were collected for molecular
autopsy. Written informed consent for a molecular autopsy was obtained
from the next-of-kin of each victim. The first-degree relatives of the
victims were referred by forensic pathologists to the study centre for
genetic counselling and recruitment into the study. Clinical history,
including personal history of arrhythmic events and history surrounding
the sudden death event of the SCD victim was collected from family
members. All recruited first-degree relatives underwent clinical
evaluation including 12-lead electrocardiogram (ECG), signal-averaged ECG,
echocardiogram, 24-hour Holter analysis and treadmill exercise testing.
Additional investigations were used only as required, such as flecainide
provocation testing (if BrS is suspected in subjects >18 years) and
targeted genetic screening (if positive molecular autopsy findings in the
index SUD victim). All first-degree relatives provided written informed
consent for these procedures and for publication of the results.
A panel of 35 genes implicated in SADS for BrS,
LQTS, short QT syndrome, CPVT, ARVC, and HCM was tested using NGS (Table
1). The NGS was performed using targeted gene capture technique on a
MiSeq Sequencing System (Illumina, Inc, San Diego [CA], United States).
Target regions of interest were restricted to the coding regions and the
10-bp flanking regions. Target rates indicated percentage of bases with a
minimum depth of coverage of 20×. Alignments to the February 2009
(GRCh37/hg19) human genome assembly and variant calls were generated using
dual pipelines, SoftGenetics NextGENe (v2.4.1) and an in-house one where
sequencing reads were aligned by Burrows-Wheeler Aligner (v0.7.5a-r405) to
hg19 genome and processed with picard-tools (v1.114). Local realignment
for indels and base quality recalibration were performed with Genome
Analysis Toolkit (GATK, v3.2). Variant calls were made with
UnifiedGenotyper (GATK v3.2). Only those genes included in the requested
gene panel were processed for variant calling. Variants identified were
annotated and analysed with VariantStudio (v2.2.1; Illumina, Inc); in
general, variants with an allele frequency of <0.1% for dominant
disorders or <1.0% for recessive disorders were reported. Pathogenic
and likely pathogenic variants were confirmed by Sanger sequencing; benign
and likely benign variants were not reported.
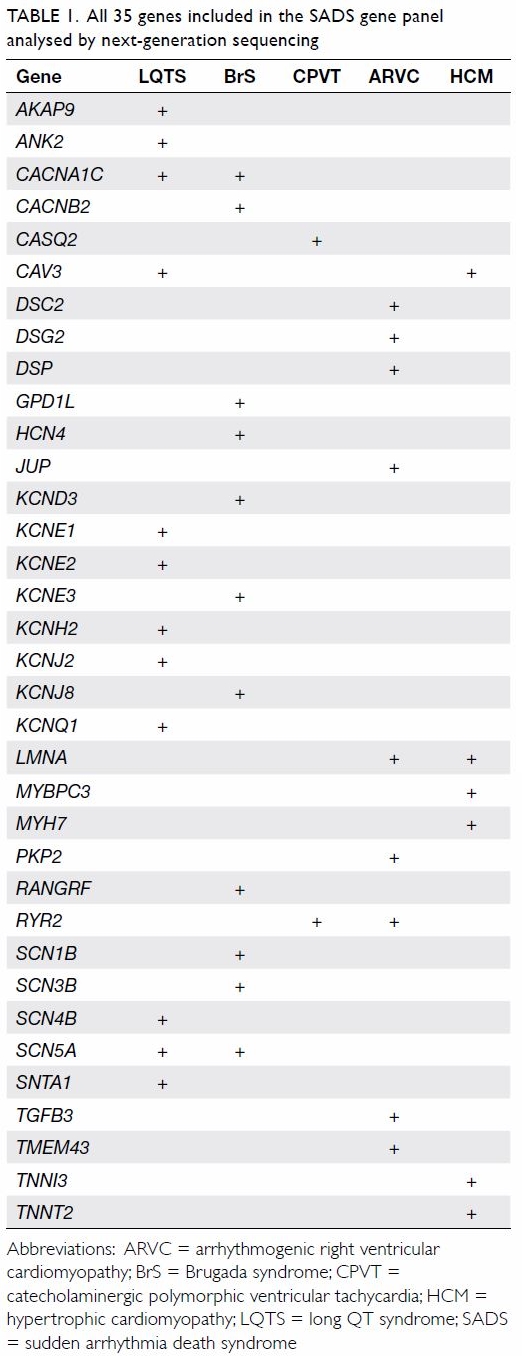
Table 1. All 35 genes included in the SADS gene panel analysed by next-generation sequencing
The genetic variant pathogenicity was established
according to the Practice Guidelines for the Interpretation and Reporting
of Unclassified Variants in Clinical Molecular Genetics by the Clinical
Molecular Genetics Society (http://
www.acgs.uk.com/media/774853/evaluation_and_reporting_of_sequence_variants_bpgs_june_2013_-_finalpdf.pdf).
Major criteria include degree of conservation, population allele
frequencies, co-segregation pattern, literature data, functional studies
and in silico prediction. The pathogenicity of novel missense variants was
analysed by PolyPhen-2, SIFT, MutationTaster and Assessing Pathogenicity
Probability in Arrhythmia by Integrating Statistical Evidence (https://cardiodb.org/APPRAISE/)
and that of novel splicing variants was by Splice Site Finder-like,
MaxEntScan, GeneSplicer and Human Splicing Finder, wherever appropriate.
Splicing variants were considered to be damaging if the score was more
than 10% lower than the wild-type prediction. Allele frequencies among
populations were as reported in the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/).
A diagnosis of SADS was established when molecular
autopsy in SCD victims identified a positive genetic variant implicated in
SADS or generally accepted clinical criteria for a particular disease were
fulfilled in the SCD victims or their first-degree relatives. The
descriptive statistics were analysed using Excel 2016 (Microsoft Corp,
Redmond [WA], United States).
Results
Five-year retrospective review of autopsy records
There were 17 187 autopsies performed during the
study period and 2748 (16%) deaths were aged 5 to 40 years. There were 420
sudden death victims. Among them, 289 (69%) were SCD (male:female
ratio=9:2). The median age was 32 years (range, 7-40 years). Coronary
artery diseases accounted for 35% of the causes of death; other causes of
death were aortic dissection (6%), myocarditis (6%), left ventricular
hypertrophy (4%), dilated cardiomyopathy (9%), other structural heart
diseases (9%), HCM (4%), ARVC (2%), and unexplained (25%).
Two-year study by conventional and molecular autopsy
There were 32 SCD victims aged 5 to 40 years
between 1 July 2014 and 30 June 2016 with a finding of either an
inheritable arrhythmogenic cardiomyopathy or no anatomical cause of death
(including other structural heart disease) on autopsy and negative
toxicology results. Eleven individuals were excluded: two who had no Hong
Kong identity card and nine whose families refused consent. Finally, 21
SCD victims (18 male, 3 female) were recruited into the study. Table
2 shows the clinical and forensic data of the 21 SCD victims. The
median age was 31 years (range, 14-39 years). From the conventional
autopsy, 18 (86%) were unrevealing; two (10%) SCD victims had ARVC and one
(5%) had HCM. The majority died during resting (48%) or sleeping (24%).
Only three SCD victims (14%) died during exercise. Three (14%) SCD victims
had family history of SCD and seven (33%) had a history of syncope. Many
(71%) of the SCD victims had unremarkable past health. Genetic analysis
showed 29% with positive heterozygous genetic variants (Table
3). Seven variants were identified in seven genes, six variants of
which were novel. The genetic background was heterogeneous without any
common mutations found. Combining conventional and molecular autopsy
findings, the cause of death in 12 (57%) still remains unknown; cause of
death was confirmed in four (19%) as ARVC, in two (10%) as BrS, and one
(5%) each in HCM, LQTS, and CPVT.
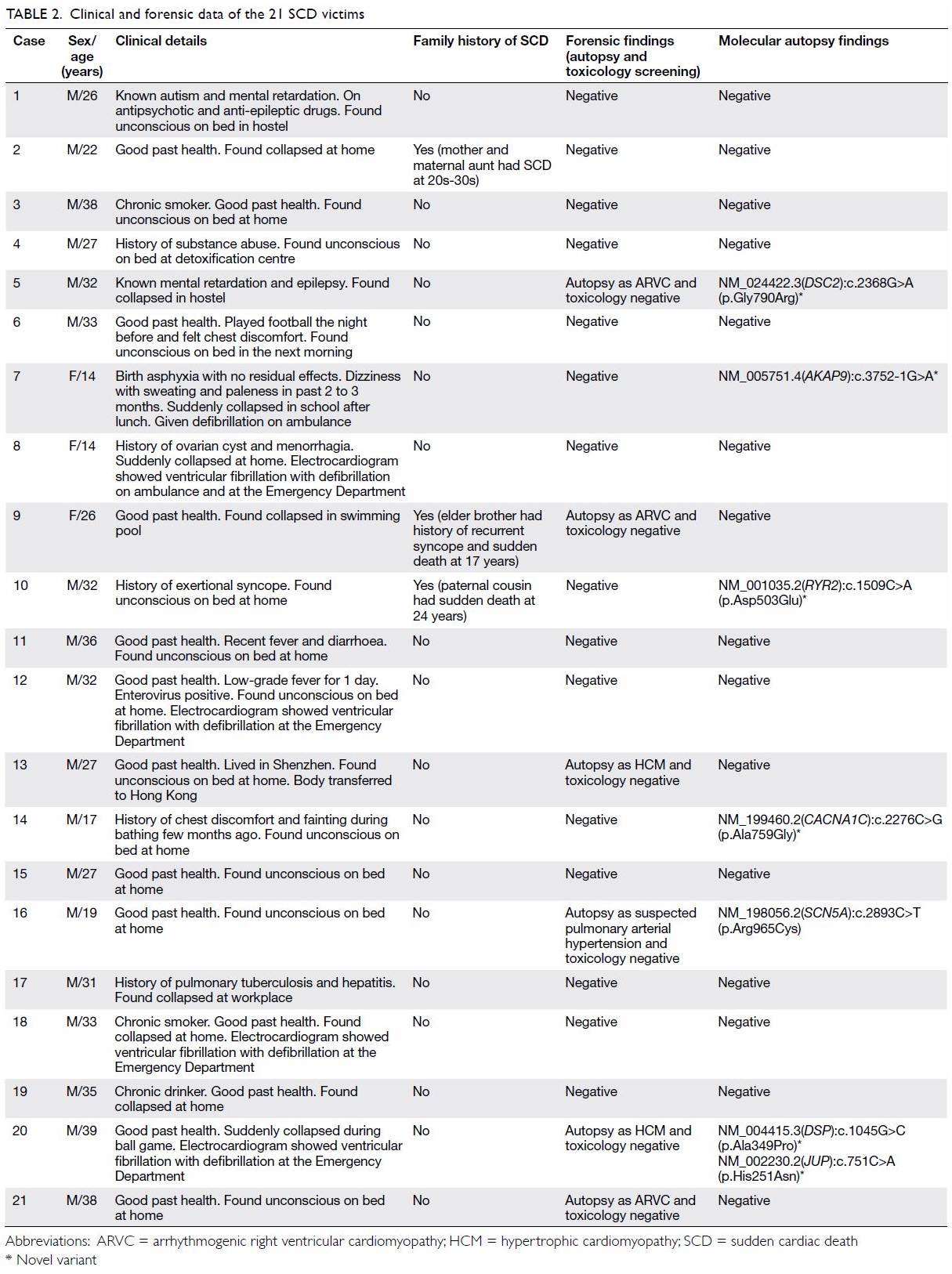
Table 2. Clinical and forensic data of the 21 SCD victims
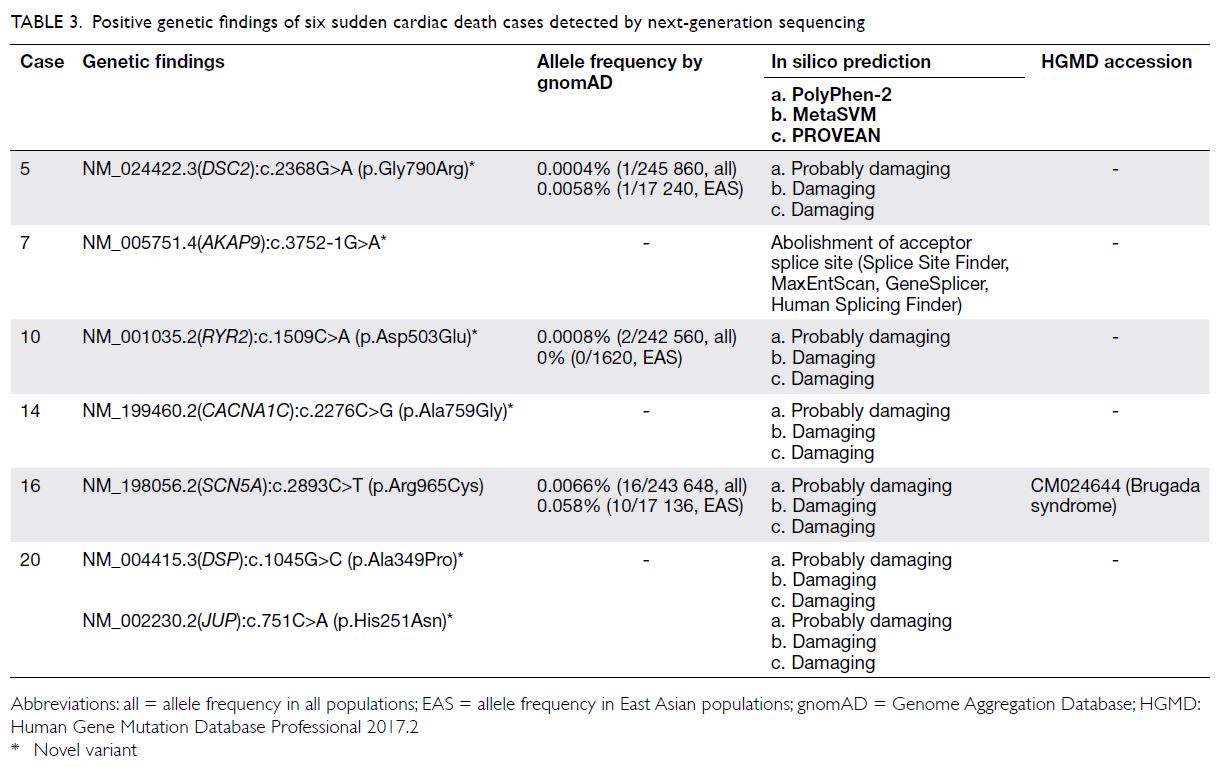
Table 3. Positive genetic findings of six sudden cardiac death cases detected by next-generation sequencing
Overall, more than half of first-degree relatives
(6 of 11 individuals) who underwent genetic testing carried the positive
genetic variants. Among them, SADS-related clinical features were detected
in three first-degree relatives from two families (cases 14 and 16). The
true clinical phenotype can sometimes be detected in surviving relatives
upon appropriate clinical evaluation, which in turn significantly aids the
interpretation of genetic findings. Cases 14 and 16 illustrate the
importance of this (Fig 1). The first-degree relatives of these two
cases showed SADS-related clinical features after cardiologist’s
assessment.
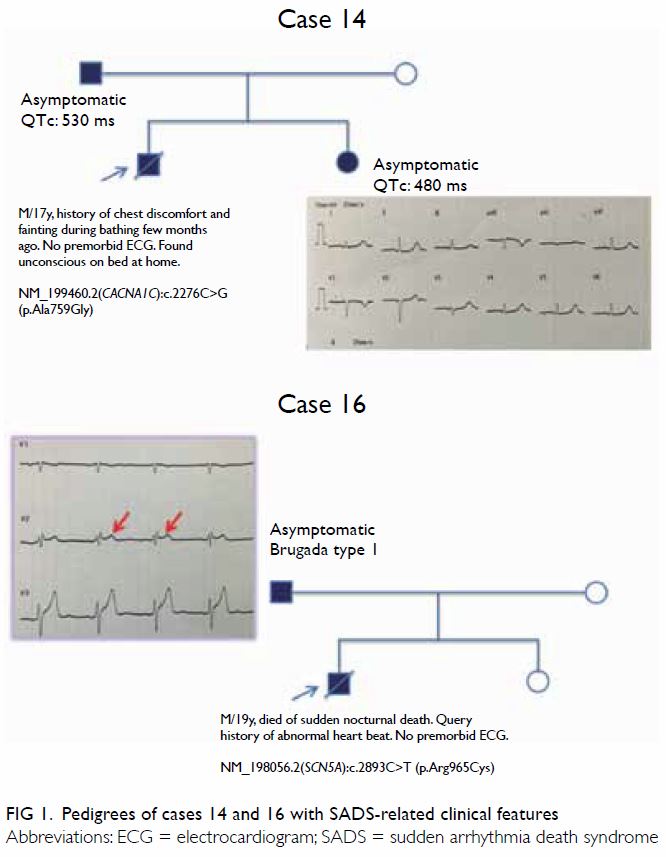
Figure 1. Pedigrees of cases 14 and 16 with SADS-related clinical features
Case 14 was a 17-year-old male victim who died of
sudden nocturnal death. He had an episode of syncope few months prior to
his death but he did not seek medical attention. Family screening by
clinical evaluation found no structural heart disease but ECG revealed
prolonged QTc interval in his asymptomatic father and sister (530 ms and
480 ms, respectively) suggesting a diagnosis of LQTS (Fig
1). Molecular autopsy found heterozygous NM_199460.2(CACNA1C):c.2276C>G
NP_955630.2:p.(Ala759Gly). This novel variant is predicted to be damaging
and absent in the gnomAD. CACNA1C is the gene encoding for the
alpha-1c subunit of the type 1 voltage-dependent calcium channel involved
in the cardiac myocyte membrane polarisation. Its mutations have been
reported in BrS and LQTS.
Case 16 was a 19-year-old male victim who also died
of sudden nocturnal death. There was no known syncope or preceding
symptoms ahead of the collapse. He had history of abnormal heart beat,
although ECG was not done. His family history was unremarkable. Autopsy
revealed pulmonary hypertension. Molecular autopsy detected heterozygous
NM_198056.2(SCN5A):c.2893C>T (p.Arg965Cys) which has been
reported as a disease causing mutation of BrS.17
18 19
The same variant has also been reported in patients with LQTS (with
digenic mutation in KCNH2 in one case).20
21 The allele frequency of this
variant (rs199473180) is reported to be 0.058% in East Asians (gnomAD). In
silico analyses by SIFT, MutationTaster, and PolyPhen-2 have revealed this
variant to be damaging, disease causing, and probably damaging,
respectively. Functional study showed that the p.(Arg965Cys) mutant led to
slower recovery from inactivation as a result of channels with a more
negative potential in steady state inactivation.18
Family screening found his father carrying the same SCN5A mutation
and a type 2 Brugada ECG pattern (Fig 1). However, no type 1 Brugada ECG feature was
revealed by a flecainide provocation test. The clinical phenotype in this
case supports the pathogenicity of this novel genetic variant.
Discussion
To elucidate the cause of SCD, a comprehensive
post-mortem evaluation is required. Figure 2 shows the combined approach using both
cardiac evaluation of surviving relatives and molecular autopsy on SUD
victims which would give a higher yield on elucidating the underlying
causes of SUD. Any history of syncope, cardiac symptoms especially related
to exertion, emotion and stress, previous ECG, circumstances of SCD
(activity at the time of death), any family history of cardiac disease,
premature or sudden death, near-arrest attack, and epilepsy should be
investigated. Patients with SADS can present with or be (incorrectly)
labelled as having epilepsy.22 23 24
25 Guidelines on post-mortem
examination of SUD in the young have been published by the Royal College
of Pathologists of Australasia (https://www.rcpa.edu.au/getattachment/89884c69-f066-411d-a3d1-
39460444db13/Guidelines-on-Autopsy-Practice.aspx). In addition to
traditional autopsy, some reports have used whole-body computed tomography
and magnetic resonance imaging to identify structural heart abnormalities
such as ARVC and HCM.26 27 However, these imaging modalities were not used in
the present study.
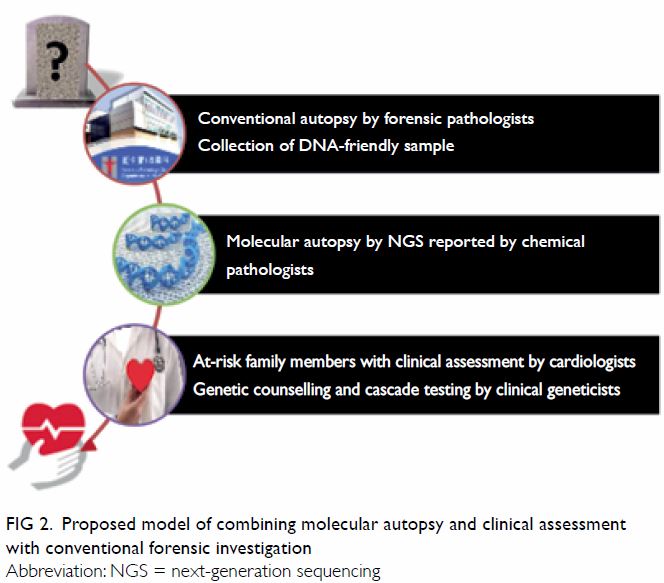
Figure 2. Proposed model of combining molecular autopsy and clinical assessment with conventional forensic investigation
Technologies applied in molecular autopsy have
changed from Sanger sequencing targeting a few major SUD-related genes to
NGS targeting an expanded gene panel of up to 200 genes.28 The former approach achieved diagnostic yields of
around 15% to 20%.8 16 29 30 31 32 With increasing throughput capacities at more
affordable costs, the large gene panel or exome approach by NGS is
becoming more appealing in molecular autopsy. A proof-of-principle
exome-wide study was carried out among 50 SUD subjects, with likely
pathogenic variants identified in 32%.33
The diagnostic yield from various NGS studies, including our own, is up to
35%, despite the different lengths of gene lists.13
34 35
36 However, the spectrum of
diseases might be different and rarer causes might be identified, because
substantial clinical suspicion is typically lacking among young SCD
victims with unremarkable premorbid history. However, a negative genetic
result does not necessarily exclude the possibility of a genetic basis for
the disease.
The major difficulty of molecular autopsy is
establishing the causation between the death and the genetic variants.
Applying molecular autopsy to the investigation of death causes involves
probabilistic rather than binary “yes or no” answers. Interpretation of
variant pathogenicity relies heavily on the characteristics of the genetic
variant, allele rarity in population frequencies, in silico predictions,
co-segregation patterns among affected family members, previous literature
data on reports of similar cases, and available functional data. This
approach follows the usual practice by which pathologists deal with
genetic reporting.
There are two main advantages of molecular autopsy
in SCD. First, molecular autopsy enables a correct diagnosis of SCD to be
established, which can bring some level of closure to the family. Findings
of a SADS-related condition can differentiate a natural cause from an
unnatural cause of death, such as the possibility of drowning precipitated
by an inherited arrhythmia during swimming. Second, the ultimate goal of
diagnosing SCD is to prevent another tragedy among family members. The
majority of SADS-related disorders are autosomal dominant inherited, and
family members are at 50% risk of inheriting the mutation, with variable
penetrance. Family pedigree for at least three generations should be
included in the extended clinical workup, and genetic counselling should
be considered for second- or higher-degree relatives wherever appropriate.
There are limitations to our study. First, SCD
victims with autopsy conducted in a public hospital were not recruited in
this study. Hence, the sample numbers may not be representative for the
whole territory. Second, no premorbid ECG findings of the victims were
available to correlate with the genetic results. Third, despite multiple
efforts, no genetic mutations were found in over two thirds of the SCD
victims in our study. Expanding the gene list may be able to increase the
yield. The associated phenotypes and pathogenesis of some genes are still
yet to be further elucidated.
Conclusion
Sudden arrhythmia death syndrome is a significant
cause of SCD in the young. This study is first of its kind in an East
Asian population and provides important data on the prevalence and types
of SADS among young SCD victims. This is also the first local feasibility
study on incorporating cardiac evaluation of surviving relatives and NGS
molecular autopsy into the conventional forensic investigations. The
interpretation of genetic variants in the context of SCD is complicated
and we recommend its analysis and reporting by qualified pathologists.
This model may be considered to cover all age-groups of SCD victims, as
well as other potential applications such as sudden unexpected death in
epilepsy, or sudden infant death syndrome. This local pilot study should
be considered an important advance in diagnosing young SCD victims in East
Asian populations.
Author contributions
All authors have made substantial contributions to
the concept or design of the study, acquisition of data, analysis or
interpretation of data, drafting of the manuscript, and critical revision
for important intellectual content. All authors had full access to the
data, contributed to the study, approved the final version for
publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of
interest.
Declaration
The 5-year review was presented at the Asia Pacific
Heart Rhythm Society Meeting on 3 to 6 October 2013, in Hong Kong. Part of
the SADS HK Study results was presented at CardioRhythm on 24 to 26
February 2017, in Hong Kong and was published (J HK Coll Cardiol
2016;24:34).
Funding/support
This work was partly funded by SADS HK Foundation
Limited, Hong Kong.
Ethics approval
The study was approved by local ethics committees
(Ethic Committee of the Department of Health (L/M 601/2013) and Kowloon
West Cluster Research Ethics Committee (KWC-REC Reference
KW/FR-13-023-67-05)). Consent was obtained from the next-of-kin of the SCD
victims and from the first-degree relatives themselves under study.
References
1. Puranik R, Chow CK, Duflou JA, Kilborn
MJ, McGuire MA. Sudden death in the young. Heart Rhythm 2005;2:1277-82. Crossref
2. Chugh SS, Reinier K, Teodorescu C, et
al. Epidemiology of sudden cardiac death: clinical and research
implications. Prog Cardiovasc Dis 2008;51:213-28. Crossref
3. Basso C, Corrado D, Thiene G.
Cardiovascular causes of sudden death in young individuals including
athletes. Cardiol Rev 1999;7:127-35. Crossref
4. Link MS. Sudden cardiac death in the
young: epidemiology and overview. Congenit Heart Dis 2017;12:597-9. Crossref
5. Chugh SS, Kelly KL, Titus JL. Sudden
cardiac death with apparently normal heart. Circulation 2000;102:649-54. Crossref
6. Corrado D, Basso C, Thiene G. Sudden
cardiac death in young people with apparently normal heart. Cardiovasc Res
2001;50:399-408. Crossref
7. de Noronha SV, Sharma S, Papadakis M,
Desai S, Whyte G, Sheppard MN. Aetiology of sudden cardiac death in
athletes in the United Kingdom: a pathological study. Heart
2009;95:1409-14. Crossref
8. Tester DJ, Medeiros-Domingo A, Will ML,
Haglund CM, Ackerman MJ. Cardiac channel molecular autopsy: insights from
173 consecutive cases of autopsy-negative sudden unexplained death
referred for postmortem genetic testing. Mayo Clin Proc 2012;87:524-39. Crossref
9. Ackerman MJ, Tester DJ, Driscoll DJ.
Molecular autopsy of sudden unexplained death in the young. Am J Forensic
Med Pathol 2001;22:105-11. Crossref
10. Tester DJ, Ackerman MJ. The role of
molecular autopsy in unexplained sudden cardiac death. Curr Opin Cardiol
2006;21:166-72. Crossref
11. Wever-Pinzon OE, Myerson M, Sherrid
MV. Sudden cardiac death in young competitive athletes due to genetic
cardiac abnormalities. Anadolu Kardiyol Derg 2009;9 Suppl 2:17-23.
12. Hellenthal N, Gaertner-Rommel A,
Klauke B, et al. Molecular autopsy of sudden unexplained deaths reveals
genetic predispositions for cardiac diseases among young forensic cases.
Europace 2017;19:1881-90. Crossref
13. Hertz CL, Christiansen SL,
Ferrero-Miliani L, et al. Next-generation sequencing of 100 candidate genes
in young victims of suspected sudden cardiac death with structural
abnormalities of the heart. Int J Legal Med 2016;130:91-102. Crossref
14. Christiansen SL, Hertz CL,
Ferrero-Miliani L, et al. Genetic investigation of 100 heart genes in
sudden unexplained death victims in a forensic setting. Eur J Hum Genet
2016;24:1797-802. Crossref
15. Semsarian C, Hamilton RM. Key role of
the molecular autopsy in sudden unexpected death. Heart Rhythm
2012;9:145-50. Crossref
16. Tester DJ, Spoon DB, Valdivia HH,
Makielski JC, Ackerman MJ. Targeted mutational analysis of the
RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a
molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin Proc
2004;79:1380-4. Crossref
17. Priori SG, Napolitano C, Gasparini M,
et al. Natural history of Brugada syndrome: insights for risk
stratification and management. Circulation 2002;105:1342-7. Crossref
18. Hsueh CH, Chen WP, Lin JL, et al.
Distinct functional defect of three novel Brugada syndrome related cardiac
sodium channel mutations. J Biomed Sci 2009;16:23. Crossref
19. Yu JH, Hu JZ, Zhou H, et al. SCN5A
mutation in patients with Brugada electrocardiographic pattern induced by
fever [in Chinese]. Zhonghua Xin Xue Guan Bing Za Zhi 2013;41:1010-4.
20. Jimmy JJ, Chen CY, Yeh HM, et al.
Clinical characteristics of patients with congenital long QT syndrome and
bigenic mutations. Chin Med J (Engl) 2014;127:1482-6.
21. Lieve KV, Williams L, Daly A, et al.
Results of genetic testing in 855 consecutive unrelated patients referred
for long QT syndrome in a clinical laboratory. Genet Test Mol Biomarkers
2013;17:553-61. Crossref
22. Medford BA, Bos JM, Ackerman MJ.
Epilepsy misdiagnosed as long QT syndrome: it can go both ways. Congenit
Heart Dis 2014;9:E135-9. Crossref
23. Partemi S, Cestèle S, Pezzella M, et
al. Loss-of-function KCNH2 mutation in a family with long QT
syndrome, epilepsy, and sudden death. Epilepsia 2013;54:e112-6. Crossref
24. Goyal JP, Sethi A, Shah VB. Jervell
and Lange-Nielson Syndrome masquerading as intractable epilepsy. Ann
Indian Acad Neurol 2012;15:145-7. Crossref
25. Tu E, Bagnall RD, Duflou J, Semsarian
C. Post-mortem review and genetic analysis of sudden unexpected death in
epilepsy (SUDEP) cases. Brain Pathol 2011;21:201-8. Crossref
26. Roberts IS, Benamore RE, Benbow EW, et
al. Post-mortem imaging as an alternative to autopsy in the diagnosis of
adult deaths: a validation study. Lancet 2012;379:136-42. Crossref
27. Puranik R, Gray B, Lackey H, et al.
Comparison of conventional autopsy and magnetic resonance imaging in
determining the cause of sudden death in the young. J Cardiovasc Magn
Reson 2014;16:44. Crossref
28. Semsarian C, Ingles J. Molecular
autopsy in victims of inherited arrhythmias. J Arrhythm 2016;32:359-65. Crossref
29. Liu C, Zhao Q, Su T, et al. Postmortem
molecular analysis of KCNQ1, KCNH2, KCNE1 and KCNE2
genes in sudden unexplained nocturnal death syndrome in the Chinese Han
population. Forensic Sci Int 2013;231:82-7. Crossref
30. Doolan A, Langlois N, Chiu C, Ingles
J, Lind JM, Semsarian C. Postmortem molecular analysis of KCNQ1
and SCN5A genes in sudden unexplained death in young Australians.
Int J Cardiol 2008;127:138-41. Crossref
31. Skinner JR, Crawford J, Smith W, et
al. Prospective, population-based long QT molecular autopsy study of
postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm
2011;8:412-9. Crossref
32. Hofman N, Tan HL, Alders M, et al.
Yield of molecular and clinical testing for arrhythmia syndromes: report
of 15 years’ experience. Circulation 2013;128:1513-21. Crossref
33. Bagnall RD, Das K J, Duflou J,
Semsarian C. Exome analysis-based molecular autopsy in cases of sudden
unexplained death in the young. Heart Rhythm 2014;11:655-62. Crossref
34. Bagnall RD, Weintraub RG, Ingles J, et
al. A prospective study of sudden cardiac death among children and young
adults. N Engl J Med 2016;374:2441-52. Crossref
35. Farrugia A, Keyser C, Hollard C, Raul
JS, Muller J, Ludes B. Targeted next generation sequencing application in
cardiac channelopathies: analysis of a cohort of autopsy-negative sudden
unexplained deaths. Forensic Sci Int 2015;254:5-11. Crossref
36. Tester DJ, Ackerman MJ. Postmortem
long QT syndrome genetic testing for sudden unexplained death in the
young. J Am Coll Cardiol 2007;49:240-6. Crossref