Hong
Kong Med J 2018 Oct;24(5):501–11 | Epub 28 Sep 2018
DOI: 10.12809/hkmj187319
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
REVIEW ARTICLE CME
Systemic lupus erythematosus: what should family
physicians know in 2018?
CC Mok, MD, FRCP
Department of Medicine, Tuen Mun Hospital, Tuen
Mun, Hong Kong
Corresponding author: Dr CC Mok (ccmok2005@yahoo.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Systemic lupus erythematosus (SLE) is a
complex multi-systemic autoimmune disease with considerable clinical and
immunological heterogeneity. Family physicians should be familiar with
the protean manifestations of SLE to aid early diagnosis and monitoring
of disease progression. The role of family physicians in SLE includes
education, counselling, psychological support, management of mild
disease, and recognition of the need for referral to other specialists
for more serious disease and complications. Surveillance of
cardiovascular risk factors and osteoporosis and advice about
vaccination and reproductive issues can be performed in the primary care
setting under close collaboration with rheumatologists and other
specialists. This review provides family physicians with the latest
classification criteria for SLE, recommendations on SLE-related health
issues, and pharmacological therapies for SLE.
Introduction
Systemic lupus erythematosus (SLE) is a
prototypical multi-systemic autoimmune disease that predominantly affects
women of childbearing age. The disease has considerable clinical and
immunological heterogeneity; no two patients with SLE are exactly alike.
The pathogenesis of SLE remains obscure, with multiple genetic,
epigenetic, hormonal, and immunopathological pathways being involved.1 The course of SLE is largely unpredictable and
characterised by periods of disease exacerbation and remission that lead
to progressive organ damage and dysfunction.2
Compared with the age- and sex-matched general population, SLE is
associated with at least a five-fold increase in mortality.3 Patients with SLE have reduced quality of life because
of multiple factors, such as organ damage, anxiety, and depression.4 5
In Hong Kong, the prevalence and annual incidence
of SLE are estimated to be 0.1% and 6.7 per 100 000 population,
respectively.6 The 15-year
cumulative survival of local Chinese patients with SLE managed in
non-academic hospitals is 86%.7
Infections, cardiovascular events, and malignancies are their most common
causes of death. Renal and musculoskeletal complications (eg, avascular
bone necrosis and osteoporotic fracture) are the most important
contributors to disease and treatment-related organ damage, respectively.
One-third of such patients lose their ability to work within 5 years after
disease onset; this is mainly attributed to musculoskeletal pain, fatigue,
anxiety/depression symptoms, and memory deterioration.8
Strategies for management of SLE by family
physicians should target early recognition and diagnosis, treatment and
monitoring of mild disease, and referral to specialists to formulate an
individualised plan based on age, disease severity, organ function, and
other medical co-morbidities.9 In
this article, the latest criteria for classification of SLE, use of
autoantibodies for diagnosis and assessment, the role of family
physicians, disease monitoring, advice on various SLE-related health
issues from a local perspective, and pharmacological treatment of the
disease will be reviewed.
Classification criteria for systemic lupus
erythematosus
The manifestations of SLE are protean, and any body
system can be involved during the course of the disease. In our experience
with 803 Hong Kong Chinese patients with SLE, the most common features are
arthritis, glomerulonephritis, facial rash, and haematological disease (Fig).7 In
primary care practice, the most frequently encountered early symptoms of
SLE include systemic upset (fatigue, fever, weight loss, loss of appetite,
and prolonged influenza-like illness), arthralgia or arthritis, facial
rash, photosensitivity, mouth sores, pleuritic chest pain, and Raynaud’s
phenomenon.6 In hospital practice,
more serious manifestations of SLE such as rapidly progressive
glomerulonephritis, pulmonary haemorrhage, cardiac tamponade, severe
cytopenia, and neuropsychiatric symptoms may be encountered.
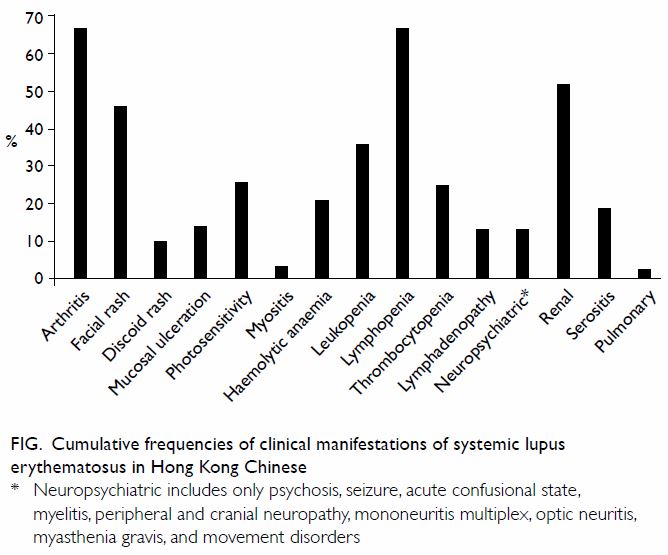
Figure. Cumulative frequencies of clinical manifestations of systemic lupus erythematosus in Hong Kong Chinese
The American College of Rheumatology (ACR)
classification criteria for SLE were established in 1982,10 and this set of criteria was revised in 1997,11 with the deletion of the LE cell phenomenon and the
addition of antiphospholipid antibodies as a criterion. A classification
of SLE is made when four or more of the 11 clinical or serological
criteria are fulfilled serially or simultaneously. However, in real life
practice, many patients with autoimmune cytopenia or hypocomplementaemia
are treated as having SLE, even though they do not fulfil the 1997
criteria.11 Moreover,
dermatological manifestations other than malar rash and discoid lesions
and neuropsychiatric manifestations other than psychosis and seizure are
not included in the criteria. Owing to these limitations, the Systemic
Lupus International Collaborating Clinics (SLICC) group revised and
validated a new set of classification criteria in 2012.12 A patient is classified as having SLE when at least
four of the 17 SLICC/ACR criteria are fulfilled. A comparison of the 1997
ACR and 2012 SLICC criteria is shown in Table 1.
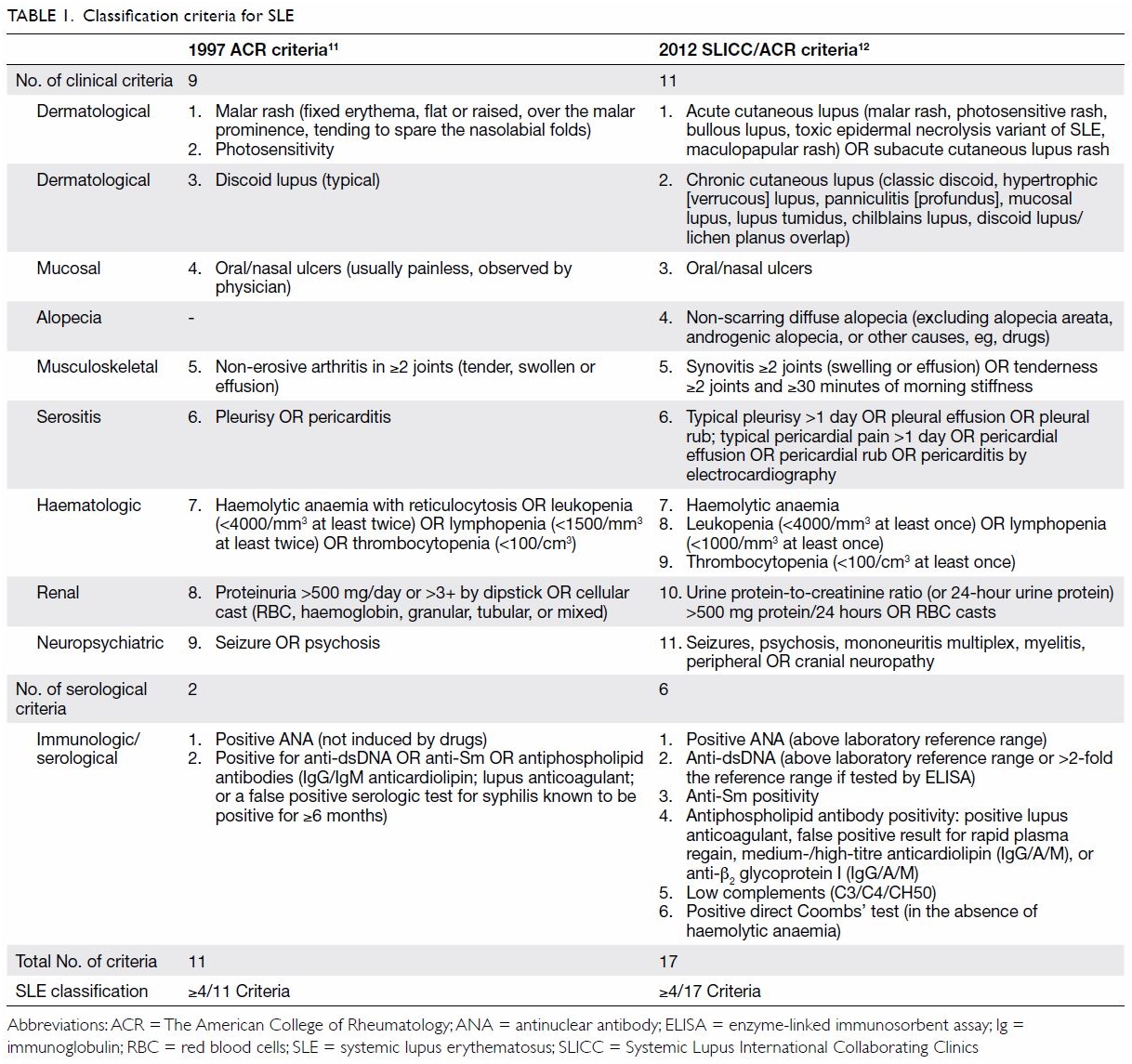
Table 1. Classification criteria for SLE
The SLICC group emphasises the absolute requirement
of at least one clinical or immunologic criterion for a classification of
SLE. Lupus malar rash and photosensitivity are no longer separated into
two criteria. There is no need to demonstrate the absence of radiological
erosion in lupus arthritis. A number of types of subacute and chronic
lupus skin lesions are included, and diffuse non-scarring alopecia
(excluding alopecia areata or other causes) is also regarded as a
criterion for SLE classification. Haemolytic anaemia,
leukopenia/lymphopenia, and thrombocytopenia are separated into three
criteria, and more neuropsychiatric features are included in addition to
psychosis and seizure. For the renal criteria, the dipstick test for urine
protein is replaced by either the protein-to-creatinine ratio (spot urine
test) or 24-hour urine protein quantification. Finally, an entity called
“stand-alone” lupus nephritis is introduced, in which a patient has
typical renal biopsy features of lupus nephritis and a positive ANA or
anti-dsDNA antibody test in the absence of other features of SLE. The
features in the SLICC criteria must be related to active SLE instead of
other causes or differential diagnoses. Validation of the SLICC criteria
has demonstrated higher sensitivity (97% vs 83%) but lower specificity
(84% vs 96%) for SLE than the 1997 ACR criteria.12
To further improve the sensitivity and specificity
of SLE classification, the ACR/EULAR (European League Against Rheumatism)
is currently validating a new set of criteria consisting of an entry
criterion and 10 domains (seven clinical and three immunologic). More
items with different weighted scores are included. Applications on mobile
devices and desktop computers will be devised to facilitate the
calculation of summed scores for classification purposes.
These classification criteria for SLE are being
developed to facilitate research and comparison among different cohorts of
patients. Although they generally have good specificity to aid diagnosis,
false positivity and negativity are bound to occur. The final diagnosis of
SLE still requires the meticulous clinical judgement of attending
physicians.
Antinuclear antibody for diagnosis of systemic lupus
erythematosus
Antinuclear antibody (ANA) is the hallmark of SLE.
Although this antibody shows extreme sensitivity for SLE (>98%), it has
low specificity. As many as 20% to 23% of normal healthy individuals test
positive for ANA, particularly older subjects.13
Other autoimmune and non-immune chronic illnesses also generate positivity
for ANA, making it grossly unsuitable as a sole diagnostic test. However,
ANA is an excellent screening test for SLE, and a negative result by
indirect immunofluorescence assay (IIFA) may virtually exclude the
diagnosis.
Antinuclear antibody is conventionally detected by
the IIFA method, which involves initial screening, serial serum dilution,
and determination of the distinct ANA staining patterns on human
epithelial cell (HEp-2) slides.14
This is the most sensitive method of ANA detection, but it is
labour-intensive and subject to inter-observer reading variability.
Although IIFA remains the gold standard of ANA detection, automated and
less laborious quantitative methods are often used by service
laboratories. Enzyme-linked immunosorbent assay is commonly used to detect
serum autoantibodies directed against antigens coated onto plates.15 As the antigens used may be derived from animal
tissues or recombinant techniques, the specificity and sensitivity of the
results for assessment of SLE vary among different commercial kits adopted
by different laboratories. In general, higher ANA titres result in more
specific predictions for SLE and related disorders. Therefore, ANA should
be interpreted in the clinical context, and a diagnosis of SLE should not
be based on a positive ANA result alone.
The dense fine speckle (DSF) pattern of ANA in IIFA
is related to autoantibodies against a 70-kDa protein (DSF70).16 Interestingly, this anti-DSF70 antibody is present in
around one-third16 17 18 19 of ANA-positive healthy subjects, in contrast to less
than 1% of ANA-positive patients with SLE and other autoimmune diseases.20 Anti-DSF70 is becoming a part of
the standard report along with the ANA result in public hospitals.
Positive results for both ANA and anti-DSF70, in the absence of other
autoantibodies such as anti-dsDNA and anti–extractable nuclear antigen
(anti-ENA), may virtually exclude SLE or an ANA-related autoimmune
disorder.
Systemic lupus erythematosus diathesis recognition
Table 2 shows a list of pointers that should alert
family physicians to consider the possibility of SLE.21 When SLE is suspected, ANA should be included in the
screening blood tests. If the patient is positive for ANA, more specific
tests such as those for anti-dsDNA, anti-ENA, antiphospholipid antibodies
(eg, anticardiolipin antibodies), and complements are needed to confirm
the diagnosis. Other relevant investigations are also needed, such as
urine analysis, cell counts, renal and liver function tests, and tests for
inflammatory markers such as erythrocyte sedimentation rate or C-reactive
protein (CRP). Diagnosis of SLE is based on a combination of compatible
clinical features and the presence of relevant immunological
abnormalities. Therefore, SLE should never be diagnosed by abnormal
antibody tests alone.

The ANA titre is not useful for monitoring of SLE
activity. The anti-dsDNA titre and complement levels (C3/4) are the
standard serological tests for disease activity evaluation (“lupus
serology”). The ENAs include a number of soluble cytoplasmic and nuclear
antigens. The six main antigens used to detect anti-ENA antibodies are Ro,
La, Sm, RNP, Scl-70, and Jo-1. The detected antibodies are associated with
certain manifestations of SLE (eg, anti-Ro with cutaneous lupus and
photosensitivity) and are relevant in pregnancy (eg, anti-Ro with
congenital heart blockage and neonatal lupus). Anti-Sm is specific to SLE
and is a criterion for its classification.11
12 As anti-ENA antibodies seldom
sero-convert over time, repeating the tests during routine follow-up is
not necessary. Table 3 summarises the assessment and monitoring of
patients with SLE and includes general advice about various health-related
issues.
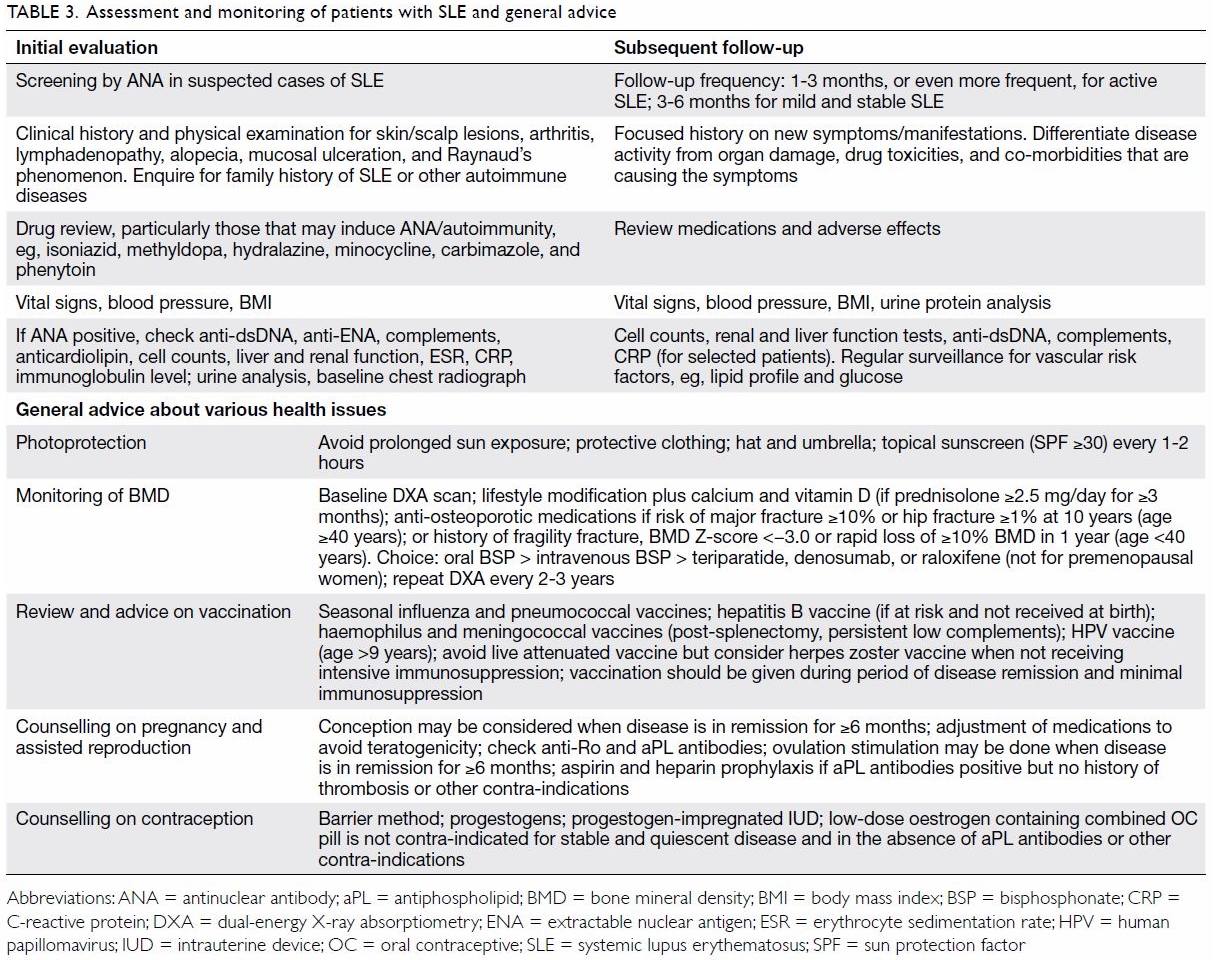
Table 3. Assessment and monitoring of patients with SLE and general advice
Role of family physicians
According to our experience with Chinese patients
with SLE, mood disorders are its most frequent psychiatric manifestations.22 The major self-reported symptoms
that lead to impaired quality of life are problems with memory and
concentration and symptoms of anxiety and depression.23 Therefore, patients with SLE should receive education
about the disease, psychological counselling, and support in the primary
care setting.
In addition to understanding the clinical
presentation of SLE for early diagnosis, trained family physicians are
able to treat and monitor mild SLE, which comprises the following
characteristics: (1) diagnosis clearly established; (2) clinically stable;
(3) absence of life-threatening manifestations; (4) stable function of
organ systems; and (5) absence of significant complications related to
disease activity or treatment. Patients with stable SLE should be followed
at intervals of 3 to 6 months. Referral to specialists is indicated for
worsening disease activity, involvement of major organs such as the
kidneys, haematological and central nervous system complications,
development of disease or treatmentrelated complications, antiphospholipid
syndrome, and advice about pregnancy, surgery, and other special
circumstances.21
Family physicians may help to monitor disease
activity and the adverse effects of drug therapies. A complete blood
count, renal function, SLE serology (anti-dsDNA and complements), and
urinary protein analysis should be performed every 3 to 6 months for
patients with stable disease. As patients with SLE are more prone to
accelerated atherosclerosis as a result of disease activity and treatment,24 surveillance for vascular risk
factors such as body mass index, blood pressure, fasting lipid profile,
and glucose should be done at regular intervals. Erythrocyte sedimentation
rate and CRP have little role in the monitoring of SLE activity and should
only be performed in patients with active synovitis undergoing specific
therapy.
Advice on photoprotection
The ultraviolet (UV) light spectrum can be divided
into UVC (100-290 nm), UVB (290-320 nm), and UVA (320-400 nm) wavelengths.
The superficial layers of the epidermis mainly absorb UVB irradiation, but
longer-wavelength UVA can also penetrate the deeper dermis. Ultraviolet
light may trigger a complicated process that includes the activation of
keratinocytes to release pro-inflammatory cytokines, chemokines, and
interferons, which may exacerbate local and systemic autoimmunity.25
Photosensitivity was poorly described in the ACR
criteria as skin rash resulting from an unusual reaction to sunlight, as
reported in patients’ history or physicians’ observation.10 11 Some
clinicians regard photosensitivity as induction or exacerbation of skin
lesions after extensive sun exposure, which also includes sunburn. Because
of the broad definition of photosensitivity, its incidence in SLE ranges
widely (27%-100% in different studies).25
The latency period between UV exposure and skin eruptions can range from
several days to 3 weeks. In addition to UV exposure, photosensitivity in
SLE can be caused by photosensitising medications and co-existing
photodermatosis.
Avoidance of excessive sunshine, particularly
during midday hours, is often advised to patients with SLE. Hats,
protective clothing, and umbrellas are effective at blocking UV light.
Ultraviolet-protective sunglasses and lip balms may also help. Topical
sunscreen is a common means of reducing UV light penetration. A sunscreen
with sun protection factor 30 absorbs/reflects 97% of UV light.26 Patients with SLE should apply sunscreen (sun
protection factor ≥30) 30 minutes before going out into the sun to all
exposed body parts and re-apply it after 1 to 2 hours if exposure is to
continue. Patients should be reminded that sunscreen does not provide 100%
protection from UV light or offer skin support for repair of photodamage.
Therefore, avoidance of unnecessary sun exposure remains the most
important behavioural modification.
Vitamin D supplementation and osteoporosis prevention
Vitamin D deficiency has recently been postulated
to be an environmental trigger for autoimmune diseases, including SLE.27 Compared with age- and sex-matched healthy subjects,
patients with SLE have significantly lower serum vitamin D levels, which
correlate inversely with disease activity.28
29 30
Vitamin D insufficiency in SLE has multiple contributing factors, which
include avoidance of UV exposure by using sunscreen, chronic kidney
disease, long-term use of medications that hamper absorption or metabolism
of vitamin D, and anti-vitamin D antibodies that may enhance plasma
clearance of vitamin D.27 Although
there is conflicting evidence regarding the efficacy of vitamin D
supplementation at alleviating clinical SLE activity, such supplementation
is recommended for prevention and treatment of glucocorticoid-induced
osteoporosis.31 According to the
updated ACR recommendations, patients receiving ≥3 months of prednisolone
(≥2.5 mg/day) should receive elemental calcium (1000-1200 mg/day) and
cholecalciferol (600-800 IU/day) along with lifestyle modification (weight
bearing exercise, cessation of smoking, balanced diet, and maintaining
optimal body weight).31 In
patients with SLE aged >40 years, who have a moderate to high risk of a
major osteoporotic (>10%) or hip fracture (>1%) within 10 years (as
assessed by the fracture risk assessment tool), oral bisphosphonates are
recommended. When oral bisphosphonates are inappropriate (eg, owing to
intolerance or contra-indication), intravenous bisphosphonates (eg,
zoledronate) are the next alternatives to be considered. Other treatment
options include teriparatide (which is costly and inconvenient to inject
daily), denosumab, and raloxifene (which has a lack of efficacy data
regarding fractures). There is a general paucity of efficacy data of these
agents in younger patients aged <40 years. The ACR recommends treatment
for moderate–to-high-risk younger patients, defined as having a previous
osteoporotic fracture; bone mineral density Z-score of <−3.0 at the hip
or spine; or rapid loss of ≥10% bone mineral density over 1 year and
continuous prednisolone treatment (≥7.5 mg/day for ≥6 months).31 The choice of drugs is the same as that for older
patients, except for raloxifene, which is not indicated in premenopausal
women or male patients.
Vaccination
Patients with SLE are prone to infections because
of the underlying immune aberrations and therapies with immunosuppressive
regimens.1 Vaccination offers the
most cost-effective method of reducing infection risk in patients with
SLE. Non-live vaccines such as influenza and pneumococcal vaccines are
generally well tolerated in SLE, although they are less immunogenic than
in age-matched individuals.32
Although there is conflicting evidence on whether influenza vaccine
exacerbates SLE activity,33
seasonal influenza vaccination according to national guidelines is
recommended.34 35 Influenza and pneumococcal vaccination is
particularly recommended for patients with SLE before rituximab therapy.
Additional vaccinations against Haemophilus influenzae and Neisseria
meningitidis are suggested for patients with functional asplenia,
splenectomy, or persistently very low complement levels.34 35 Hepatitis
B vaccination can be safely administered to patients with SLE who are at
risk of infection if it was not given at birth. Female patients with SLE
are more prone to persistent genital human papillomavirus (HPV) infection,
which predisposes them to cervical cancers. The HPV vaccine is recommended
for patients with SLE, preferably prior to the beginning of sexual
activity. There is no evidence of increased SLE flares after
administration of the quadrivalent or bivalent HPV vaccines.36 In Hong Kong, the quadrivalent and nonavalent HPV
vaccine is licensed for female and male patients aged ≥9 years.37 Non-live vaccines should be given to patients with
SLE during periods of disease quiescence and minimal immunosuppression.
Live attenuated vaccines are generally not
recommended for individuals who are heavily immunocompromised because of
the risk of disseminated infections. Of relevance is the live attenuated
herpes zoster vaccine, which has been licensed for patients aged >50
years. Patients with SLE are particularly prone to herpes zoster
reactivation, with a pooled relative risk of 2.10 compared with the age-
and sex-matched general population.38
According to the United States Advisory Committee on Immunization
Practices, herpes zoster vaccine should not be given to individuals who
are receiving heavy immunosuppressive therapies, such as prednisolone
(>20 mg/day for ≥2 weeks), methotrexate (≥0.4 mg/kg/week), and
azathioprine (≥3.0 mg/kg/day).39
However, in view of the high incidence of herpes zoster in patients with
SLE, herpes zoster vaccine should be considered in those who have stable
and remitted disease that does not require intense immunosuppression.34 35 The
herpes zoster vaccine has been administered safely to SLE patients without
subsequent development of herpetiform lesions or disease flares.40
Pregnancy counselling, assisted reproduction, and
contraception
The fertility of patients with SLE is preserved,
unless they develop chronic kidney disease or have been treated with
cyclophosphamide. Patients with SLE should not be discouraged regarding
pregnancy, provided that their disease has been under good control for at
least 6 to 12 months.41 The
outcomes of pregnancies have improved for patients with SLE in the past
few decades as a result of better risk stratification, pre-conception
counselling, and close multidisciplinary surveillance. However, the rates
of pregnancy loss, preterm birth, pre-eclampsia, and intrauterine growth
retardation remain higher in pregnancies of patients with SLE than in
those of patients without.41 The
main risk factors for poor maternal and fetal outcomes in pregnancies of
patients with SLE are active disease at conception (particularly
nephritis), the presence of strongly positive antiphospholipid antibodies
(or a history of obstetric antiphospholipid syndrome), and a history of
lupus nephritis.42 Some
medications such as cyclophosphamide, mycophenolate mofetil, leflunomide,
and angiotensin-converting enzyme inhibitors/angiotensin receptor blockers
are teratogenic. High-dose glucocorticoid treatment may lead to
intrauterine growth retardation and premature delivery. The risk of
congenital heart blockage in anti-Ro-positive mothers with SLE is
approximately 1% to 2%.42 Close
liaison with obstetricians and paediatricians for monitoring of the
cardiovascular status of the fetus during pregnancy and assessment of
neonatal lupus syndrome is needed. In general, SLE patients with ≥6 months
of disease remission who are in good general health may consider
conception. Referral to specialists for adjustment of medications and
prophylactic heparin/aspirin (in case of obstetric antiphospholipid
syndrome) is needed.
The use of assisted reproductive technology is
increasing. Despite increases in disease flares and thrombosis after
hormonal ovulation stimulation,43
the current recommendation is to individualise the risk of these
procedures in patients with SLE.44
Assisted reproductive technology procedures should be discouraged in
female SLE patients who have active disease, severe renal insufficiency,
serious valvulopathy or coronary heart disease, poorly controlled
hypertension, history of major thrombotic events, or antiphospholipid
syndrome.44 Counselling should
also be given about other serious adverse effects of assisted reproductive
technology procedures, such as ovarian hyperstimulation. In patients with
SLE who are positive for antiphospholipid antibodies and have no history
of thrombosis, aspirin and heparin prophylaxis is recommended during these
procedures.45 Similar to naturally
achieved pregnancies, the SLE of candidates for assisted reproductive
technology should have been quiescent for ≥6 months.46
Patients with SLE should be counselled about
contraception methods. Barrier methods are generally safe.
Oestrogen-containing oral contraceptive pills were discouraged in the
past. However, in a randomised double-blind placebo-controlled trial, a
combination of oral contraceptive pills was not shown to increase SLE
disease flares or thrombosis after 12 months’ administration as compared
with placebo in patients with stable SLE and no antiphospholipid
antibodies.47 Another randomised
controlled trial did not reveal a difference in disease flares or adverse
events in 12 months among patients with SLE who were assigned to receive
combined oral contraceptive pill, intrauterine device, and
progestogen-only pills for contraception.48
Thus, patients with stable SLE and no antiphospholipid antibodies or other
contra-indications may use low-dose oestrogen oral contraceptive pills if
they want to adopt a more reliable contraceptive method. When oral
contraceptive pills are not appropriate, progestogens and intrauterine
devices can be offered to patients with SLE as alternatives.44 Progestogen-impregnated intrauterine devices have the
advantage of reducing the incidence of dysmenorrhoea and irregular vaginal
bleeding.44
Conventional and novel therapeutics for systemic lupus
erythematosus
Hydroxychloroquine is an antimalarial drug that
exhibits immune-modulatory properties in addition to antithrombotic and
lipid- and glucose-lowering properties.49
Hydroxychloroquine is mainly indicated for skin, joint, and serosal
manifestations of SLE and has a glucocorticoid-sparing effect. The drug is
compatible with pregnancy and breastfeeding and is relatively safe to be
prescribed and monitored by trained family physicians. Allergy and acute
ocular and neuromuscular toxicity are rare adverse drug reactions. Chronic
use of hydroxychloroquine may lead to retinopathy, with the main risk
factors being older age, pre-existing liver and renal dysfunction, higher
daily dose, and longer duration of therapy.50
51 Early recognition of this
adverse drug reaction is essential to minimise damage to vision. Referral
to an ophthalmologist for baseline examination and regular retinopathy
surveillance is recommended.50
In a recent study, 2361 patients received
hydroxychloroquine for >5 years. In that study, the risk of retinopathy
was <1% in the first 5 years and <2% in 10 years when the daily dose
was <5 mg/ kg of real body weight.51
The risk of retinopathy increased sharply to 20% after 20 years. The daily
dose of hydroxychloroquine was the most critical factor for the
retinopathy risk, which correlated better with real rather than ideal body
weight. The American Academy of Ophthalmology recommends a maximum daily
hydroxychloroquine dose of <5.0 mg/kg of real weight to minimise
retinal toxicity.50 A baseline
ophthalmologic examination within the first year of commencement of drug
administration is recommended, and annual screening should start after 5
years of exposure in patients using a lower dosage and without major risk
factors. Patients with major risk factors for retinopathy (older age,
renal or liver dysfunction, or pre-existing macular or retinal disease)
should be screened annually if not more frequently.50
Short courses of non-steroidal anti-inflammatory
drugs (NSAIDs) are indicated for control of SLE symptoms such as
arthritis, myalgia, serositis, and fever. The risk of allergic and skin
reactions, aseptic meningitis, and renal and liver toxicity is increased
in SLE patients, despite their younger age. Ovulation may be affected by
NSAIDs, and they should be used cautiously during pregnancy. Patients with
SLE who have renal insufficiency, bleeding tendency, and pre-existing
coronary heart disease should avoid NSAIDs. Except for their lower risk of
gastrointestinal toxicity, selective Cox II inhibitors share similar
renal, hepatological, and neurological adverse effects with non-selective
Cox inhibitors.52 Among the
NSAIDs, naproxen appears to be associated with the lowest risk of
cardiovascular events and is the preferred NSAID for patients with
multiple cardiovascular risk factors.53
Diclofenac is associated with the highest risk and should be avoided in
these patients. A recent randomised controlled trial reported that
celecoxib was non-inferior to ibuprofen or naproxen with regard to
cardiovascular safety in patients with rheumatoid arthritis and
osteoarthritis.54 The lowest
effective dose of NSAIDs should be used, and their indications should be
periodically reviewed. Monitoring of fluid status, kidney function, liver
transaminases, and blood pressure is necessary.
Glucocorticoids and a number of non-glucocorticoid
immunosuppressive agents are often used to treat more serious organ
manifestations of SLE. Systemic glucocorticoids are a major cause of
treatment-related organ damage in patients with SLE and contribute
significantly to mortality and co-morbidities.7
55 Therefore, the use of systemic
glucocorticoids in SLE has to be fully justified, judicious, and closely
monitored. Other treatment modalities used for severe SLE include
intravenous immunoglobulin and plasmapheresis. A biological agent called
belimumab has recently been approved for mild to moderate SLE
manifestations that are refractory to standard therapies.56 Although rituximab has not been proven to be more
effective than placebo in randomised controlled trials, it is often used
off-label for refractory lupus manifestations.1
Many other biological and targeted synthetic agents are being tested in
patients with SLE. While it is outside the scope of this review to
describe these therapies in detail, they are summarised in Table
4 for quick reference.
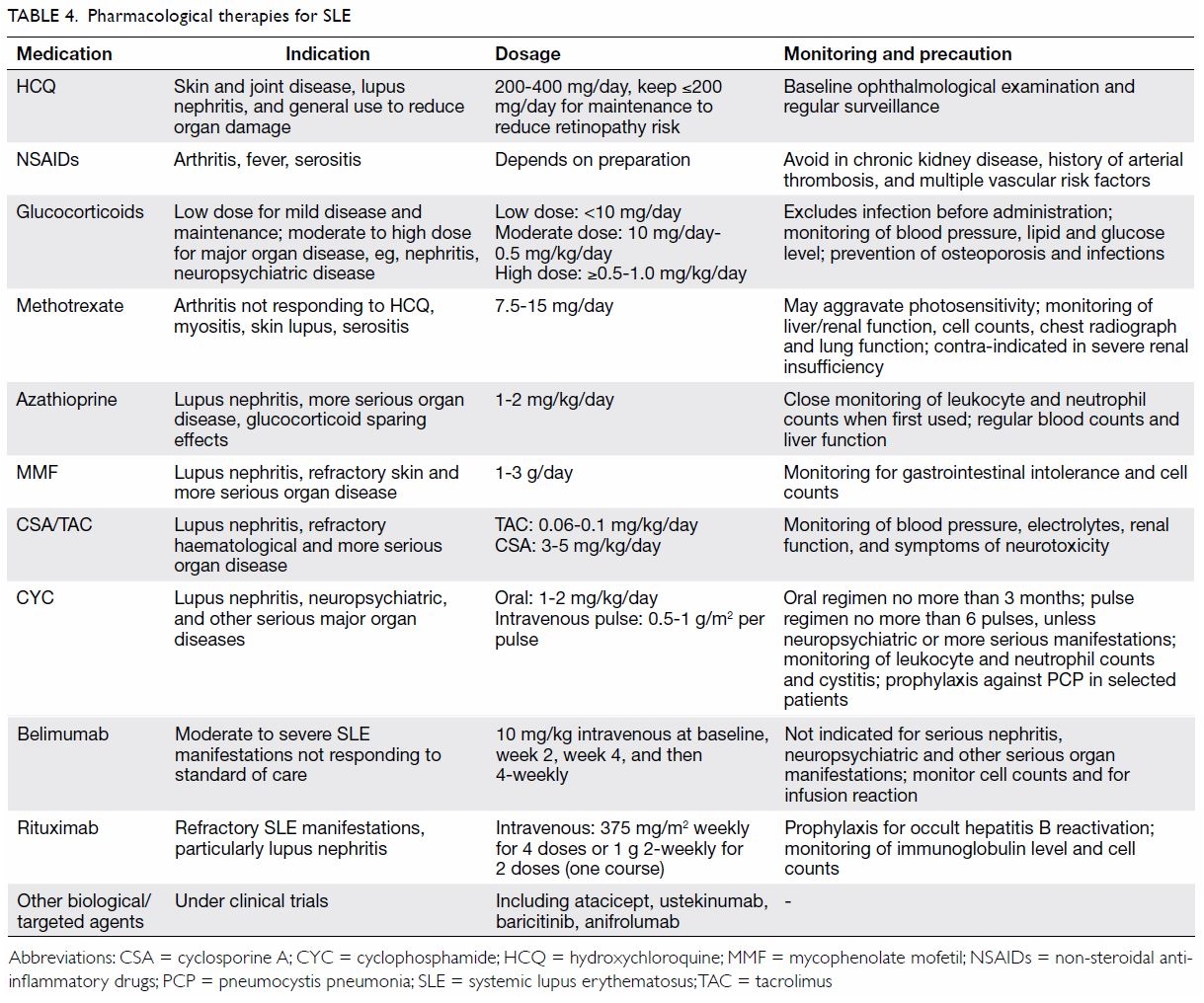
Table 4. Pharmacological therapies for SLE
Conclusions
Systemic lupus erythematosus is a prototypical
autoimmune disease that affects primarily young women of reproductive age.
The new SLICC classification has expanded the clinical and serological
criteria for its classification. Systemic lupus erythematosus should never
be diagnosed based solely on positive test results for antibodies,
particularly ANA, which is highly non-specific and should be interpreted
in conjunction with clinical signs and symptoms. In view of the disease’s
multisystemic involvement, holistic care is necessary to formulate
treatment plans for individual patients. Family physicians play an
important role in establishing an early diagnosis, treatment and
monitoring of mild disease, and making referrals to specialists when
appropriate. Education, counselling, and psychological support are equally
important to improve treatment adherence and alleviate mood symptoms.
General advice about photoprotection, vaccination, prevention of
osteoporosis, and reproductive issues may be given in the primary care
setting. Hydroxychloroquine is a relatively safe drug that can be
commenced and monitored by family physicians. For patients with stable
SLE, screening for cardiovascular risk factors and osteoporosis may also
be performed periodically in family clinics.
Author contributions
The author has made substantial contributions to
the concept or design, acquisition of data, analysis or interpretation of
data, drafting of the article, and critical revision for important
intellectual content.
Declaration
The author has disclosed no conflicts of interest.
The author had full access to the data, contributed to the study, approved
the final version for publication, and take responsibility for its
accuracy and integrity.
References
1. Mok CC. Biological and targeted
therapies of systemic lupus erythematosus: evidence and the state of the
art. Expert Rev Clin Immunol 2017;13:677-92. Crossref
2. Mok CC. Epidemiology and survival of
systemic lupus erythematosus in Hong Kong Chinese. Lupus 2011;20:767-71. Crossref
3. Mok CC, Kwok CL, Ho LY, Chan PT, Yip SF.
Life expectancy, standardized mortality ratios, and causes of death in six
rheumatic diseases in Hong Kong, China. Arthritis Rheum 2011;63:1182-9. Crossref
4. Mok CC, Kosinski M, Ho LY, Chan KL,
Jolly M. Validation of the LupusPRO in Chinese patients from Hong Kong
with systemic lupus erythematosus. Arthritis Care Res (Hoboken)
2015;67:297-304. Crossref
5. Mok CC, Ho LY, Cheung MY, Yu KL, To CH.
Effect of disease activity and damage on quality of life in patients with
systemic lupus erythematosus: a 2-year prospective study. Scand J
Rheumatol 2009;38:121-7. Crossref
6. Mok CC. Emerging biological therapies
for systemic lupus erythematosus. Expert Opin Emerg Drugs 2014;19:303-22.
Crossref
7. Mok CC, Tse SM, Chan KL, Ho LY. Effect
of immunosuppressive therapies on survival of systemic lupus
erythematosus: a propensity score analysis of a longitudinal cohort. Lupus
2018;27:722-7. Crossref
8. Mok CC, Cheung MY, Ho LY, Yu KL, To CH.
Risk and predictors of work disability in Chinese patients with systemic
lupus erythematosus. Lupus 2008;17:1103-7. Crossref
9. Lam NC, Ghetu MV, Bieniek ML. Systemic
lupus erythematosus: primary care approach to diagnosis and management. Am
Fam Physician 2016;94:284-94.
10. Tan EM, Cohen AS, Fries JF, et al. The
1982 revised criteria for the classification of systemic lupus
erythematosus. Arthritis Rheum 1982;25:1271-7. Crossref
11. Hochberg MC. Updating the American
College of Rheumatology revised criteria for the classification of
systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. Crossref
12. Petri M, Orbai AM, Alarcón GS, et al.
Derivation and validation of the Systemic Lupus International
Collaborating Clinics classification criteria for systemic lupus
erythematosus. Arthritis Rheum 2012;64:2677-86. Crossref
13. Pisetsky DS. Antinuclear antibody
testing—misunderstood or misbegotten? Nat Rev Rheumatol 2017;13:495-502. Crossref
14. Rigon A, Infantino M, Merone M, et al.
The inter-observer reading variability in anti-nuclear antibodies indirect
(ANA) immunofluorescence test: A multicenter evaluation and a review of
the literature. Autoimmun Rev 2017;16:1224-9.Crossref
15. Emlen W, O’Neill L. Clinical
significance of antinuclear antibodies: comparison of detection with
immunofluorescence and enzyme-linked immunosorbent assays. Arthritis Rheum
1997;40:1612-8. Crossref
16. Dellavance A, Viana VS, Leon EP, Bonfa
ES, Andrade LE, Leser PG. The clinical spectrum of antinuclear antibodies
associated with the nuclear dense fine speckled immunofluorescence
pattern. J Rheumatol 2005;32:2144-9.
17. Infantino M, Meacci F, Grossi V, et
al. The clinical impact of anti-DFS70 antibodies in undifferentiated
connective tissue disease: case reports and a review of the literature.
Immunol Res 2017;65:293-5. Crossref
18. Mahler M, Hanly JG, Fritzler MJ.
Importance of the dense fine speckled pattern on HEp-2 cells and
anti-DFS70 antibodies for the diagnosis of systemic autoimmune diseases.
Autoimmun Rev 2012;11:642-5. Crossref
19. Mariz HA, Sato EI, Barbosa SH,
Rodrigues SH, Dellavance A, Andrade LE. Pattern on the antinuclear
antibody-HEp-2 test is a critical parameter for discriminating antinuclear
antibody-positive healthy individuals and patients with autoimmune
rheumatic diseases. Arthritis Rheum 2011;63:191-200. Crossref
20. Seelig CA, Bauer O, Seelig HP.
Autoantibodies against DFS70/LEDGF exclusion markers for systemic
autoimmune rheumatic diseases (SARD). Clin Lab 2016;62:499-517. Crossref
21. Mok CC. Systemic lupus erythematosus:
when to refer? HK Pract 2002;24:444-9.
22. Mok CC, To CH, Mak A. Neuropsychiatric
damage in Southern Chinese patients with systemic lupus erythematosus.
Medicine (Baltimore) 2006;85:221-8. Crossref
23. Mok CC, Ho LY, Tse SM, Chan KL.
Prevalence of remission and its effect on damage and quality of life in
Chinese patients with systemic lupus erythematosus. Ann Rheum Dis
2017;76:1420-5. Crossref
24. Mok CC. Accelerated atherosclerosis,
arterial thromboembolism, and preventive strategies in systemic lupus
erythematosus. Scand J Rheumatol 2006;35:85-95. Crossref
25. Kuhn A, Wenzel J, Bijl M. Lupus
erythematosus revisited. Semin Immunopathol 2016;38:97-112. Crossref
26. Obermoser G, Zelger B. Triple need for
photoprotection in lupus erythematosus. Lupus 2008;17:525-7. Crossref
27. Mok CC. Vitamin D and systemic lupus
erythematosus: an update. Expert Rev Clin Immunol 2013;9:453-63. Crossref
28. Shahin D, El-Farahaty RM, Houssen ME,
et al. Serum 25-OH vitamin D level in treatment-naïve systemic lupus
erythematosus patients: relation to disease activity, IL-23 and IL-17.
Lupus 2017;26:917-26. Crossref
29. Mok CC, Birmingham DJ, Leung HW,
Hebert LA, Song H, Rovin BH. Vitamin D levels in Chinese patients with
systemic lupus erythematosus: relationship with disease activity, vascular
risk factors and atherosclerosis. Rheumatology (Oxford) 2012;51:644-52. Crossref
30. Amital H, Szekanecz Z, Szücs G, et al.
Serum concentrations of 25-OH vitamin D in patients with systemic lupus
erythematosus (SLE) are inversely related to disease activity: is it time
to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis
2010;69:1155-7. Crossref
31. Buckley L, Guyatt G, Fink HA, et al.
2017 American College of Rheumatology Guideline for the prevention and
treatment of glucocorticoid-induced osteoporosis. Arthritis Rheumatol
2017;69:1521-37. Crossref
32. Pugès M, Biscay P, Barnetche T, et al.
Immunogenicity and impact on disease activity of influenza and
pneumococcal vaccines in systemic lupus erythematosus: a systematic
literature review and meta-analysis. Rheumatology (Oxford)
2016;55:1664-72. Crossref
33. Murdaca G, Orsi A, Spanò F, et al.
Vaccine-preventable infections in systemic lupus erythematosus. Hum Vaccin
Immunother 2016;12:632-43. Crossref
34. van Assen S, Agmon-Levin N, Elkayam O,
et al. EULAR recommendations for vaccination in adult patients with
autoimmune inflammatory rheumatic diseases. Ann Rheum Dis 2011;70:414-22.
Crossref
35. Heijstek MW, Ott de Bruin LM, Bijl M,
et al. EULAR recommendations for vaccination in paediatric patients with
rheumatic diseases. Ann Rheum Dis 2011;70:1704-12. Crossref
36. Mok CC, Ho LY, Fong LS, To CH.
Immunogenicity and safety of a quadrivalent human papillomavirus vaccine
in patients with systemic lupus erythematosus: a case-control study. Ann
Rheum Dis 2013;72:659-64. Crossref
37. Cervical screening programme,
Department of Health, Hong Kong SAR Government. Available from:
https://www.cervicalscreening.gov.hk/english/hum/hum_ccv.html#4. Accessed
1 May 2018.
38. Kawai K, Yawn BP. Risk factors for
herpes zoster: a systematic review and meta-analysis. Mayo Clin Proc
2017;92:1806-21. Crossref
39. Harpaz R, Ortega-Sanchez IR, Seward
JF; Advisory Committee on Immunization Practices (ACIP), Centers for
Disease Control and Prevention. Prevention of herpes zoster:
recommendations of the Advisory Committee on Immunization Practices
(ACIP). MMWR Recomm Rep 2008;57:1-30.
40. Guthridge JM, Cogman A, Merrill JT, et
al. Herpes zoster vaccination in SLE: a pilot study of immunogenicity. J
Rheumatol 2013;40:1875-80. Crossref
41. Fischer-Betz R, Specker C. Pregnancy
in systemic lupus erythematosus and antiphospholipid syndrome. Best Pract
Res Clin Rheumatol 2017;31:397-414. Crossref
42. Peart E, Clowse ME. Systemic lupus
erythematosus and pregnancy outcomes: an update and review of the
literature. Curr Opin Rheumatol 2014;26:118-23. Crossref
43. Orquevaux P, Masseau A, Le Guern V, et
al. In vitro fertilization in 37 women with systemic lupus erythematosus
or antiphospholipid syndrome: a series of 97 procedures. J Rheumatol
2017;44:613-8. Crossref
44. Andreoli L, Crisafulli F, Tincani A.
Pregnancy and reproductive aspects of systemic lupus erythematosus. Curr
Opin Rheumatol 2017;29:473-9. Crossref
45. Andreoli L, Bertsias GK, Agmon-Levin
N, et al. EULAR recommendations for women’s health and the management of
family planning, assisted reproduction, pregnancy and menopause in
patients with systemic lupus erythematosus and/or antiphospholipid
syndrome. Ann Rheum Dis 2017;76:476-85. Crossref
46. Levine AB, Lockshin MD. Assisted
reproductive technology in SLE and APS. Lupus 2014;23:1239-41. Crossref
47. Petri M, Kim MY, Kalunian KC, et al.
Combined oral contraceptives in women with systemic lupus erythematosus. N
Engl J Med 2005;353:2550-8. Crossref
48. Sánchez-Guerrero J, Uribe AG,
Jiménez-Santana L, et al. A trial of contraceptive methods in women with
systemic lupus erythematosus. N Engl J Med 2005;353:2539-49. Crossref
49. Ponticelli C, Moroni G.
Hydroxychloroquine in systemic lupus erythematosus (SLE). Expert Opin Drug
Saf 2017;16:411-9. Crossref
50. Marmor MF, Kellner U, Lai TY, Melles
RB, Mieler WF; American Academy of Ophthalmology. Recommendations on
screening for chloroquine and hydroxychloroquine retinopathy (2016
revision). Ophthalmology 2016;123:1386-94. Crossref
51. Melles RB, Marmor MF. The risk of
toxic retinopathy in patients on long-term hydroxychloroquine therapy.
JAMA Ophthalmol 2014;132:1453-60. Crossref
52. Ostensen M, Villiger PM. Nonsteroidal
anti-inflammatory drugs in systemic lupus erythematosus. Lupus
2000;9:566-72. Crossref
53. Pelletier JP, Martel-Pelletier J,
Rannou F, Cooper C. Efficacy and safety of oral NSAIDs and analgesics in
the management of osteoarthritis: evidence from real-life setting trials
and surveys. Semin Arthritis Rheum 2016;45(4 Suppl):S22-7. Crossref
54. Nissen SE, Yeomans ND, Solomon DH, et
al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for
arthritis. N Engl J Med 2016;375:2519-29. Crossref
55. Tarr T, Papp G, Nagy N, Cserép E,
Zeher M. Chronic high-dose glucocorticoid therapy triggers the development
of chronic organ damage and worsens disease outcome in systemic lupus
erythematosus. Clin Rheumatol 2017;36:327-33. Crossref
56. Hahn BH. Belimumab for systemic lupus
erythematosus. N Engl J Med 2013;368:1528-35. Crossref