Hong
Kong Med J 2018 Oct;24(5):451–9 | Epub 28 Sep 2018
DOI: 10.12809/hkmj187260
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Genetic profile and clinical application of chromosomal
microarray in children with intellectual disability in Hong Kong
Purdy YT Chan, FHKCPaed, FHKAM (Paediatrics)1;
HM Luk, MD (HK), FHKAM (Paediatrics)2; Florence MY Lee,
FHKCPaed, FHKAM (Paediatrics)1; Ivan FM Lo, FHKCPaed, FHKAM
(Paediatrics)2
1 Child Assessment Service, Department
of Health, Hong Kong
2 Clinical Genetic Service, Department
of Health, Hong Kong
Corresponding author: Dr Ivan FM Lo (con_cg@dh.gov.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Introduction: Chromosomal
microarray (CMA) is recommended as a first-tier genetic investigation
for intellectual disability (ID), developmental delay, or autism
spectrum disorder due to its higher diagnostic yield with respect to
conventional karyotyping. The aim of the present study was to
investigate the genetic profile and diagnostic yield of CMA in children
with moderate, severe and profound ID.
Methods: A pilot cross-sectional
study was performed by the Child Assessment Service and the Clinical
Genetic Service in Hong Kong from July 2016 to June 2017. Children with
unexplained ID were recruited for CMA testing by an expedited referral
pathway. Children who were existing clients of the Clinical Genetic
Service were also recruited.
Results: Of 225 children
included in this study, 68 (30.2%) had genetic diagnoses. Among the 138
children who underwent CMA testing, 53 (38%) children were referred to
the Clinical Genetic Service by the expedited referral pathway. The
respective diagnostic yields of CMA in moderate, severe, and profound ID
were 8.7%, 17.6%, and 23.5% (P<0.05). Children with dysmorphic
features demonstrated a much higher yield from CMA (45.8% vs 4.4%,
P<0.05).
Conclusion: The overall
diagnostic yield (11.6%) of CMA in this cohort is comparable with that
of other international cohorts. This further supports the use of CMA as
a first-tier genetic investigation for children with ID, developmental
delay, or autism spectrum disorder, particularly for those with severe
disease.
New knowledge added by this study
- Approximately one-third of children with more severe forms of intellectual disability exhibited a genetic condition, as determined by chromosomal microarray.
- The diagnostic yield of chromosomal microarray testing increases with the severity of intellectual disability, and with the severity of dysmorphic features.
- The expedited mechanism, if extended to younger children with developmental delay (with or without autism spectrum disorder), may avoid unnecessary investigations in children, improve the efficiency of service delivery, and reduce societal cost.
Introduction
Intellectual disability (ID) is estimated to affect
1% to 3% of the population in Western societies.1
It is almost two-fold greater in prevalence in low-and middle-income
countries, compared with high-income countries. Importantly, the General
Household Survey in 2014 showed the prevalence rate of ID to be
approximately 1.0% to 1.4% in Hong Kong.2
Intellectual disability is defined as ‘significant
limitations both in intellectual functioning and in adaptive behaviour, as
expressed in conceptual, social, and practical adaptive skills’.3 These difficulties are evident above the age of 18; ID
is indicated by an intelligence quotient (IQ) of approximately two
standard deviations (SD) or more below the population mean (IQ ≤70) on the
IQ test.
Developmental delay (DD) describes the
developmental level of a child, typically <5 years old, who is
substantially below the average standard of his peers. Global DD is
defined as a significant delay in two or more domains: gross motor, fine
motor, language, cognitive, social, or activities of daily living.
Significant delay refers to scores >2 SD below the mean on
norm-referenced age-appropriate developmental tests.4
In 2015 and 2016, more than 3000 children per year
were diagnosed with DD by the Child Assessment Service (CAS). A study
conducted in 2003 to 2004 showed that 80% of children with significant
delay and 30% of children with borderline delay were later confirmed to
exhibit ID at an older age.5
Overall, 30% of children with ID had a co-morbid diagnosis of autism
spectrum disorder (ASD).
The aetiology of ID is complex. While milder forms
of ID are suspected to typically result from the interplay of genetic and
environmental factors,6 biological
causes, particularly genetic causes, are often identified in children with
significant cognitive delays (IQ <50).4
Rauch et al7 studied 670 subjects,
generally <6 years of age, with ID: in 39.5%, ID was related to a
genetic cause; in 1.3%, it was due to an acquired or environmental cause;
and in 50% to 60%, it did not exhibit a known aetiology.
Chromosomal microarray (CMA) or array comparative
genomic hybridisation, is recommended by many international professional
organisations as a first-tier genetic investigation for children with
unexplained DD, ID, or ASD.4 8 9 Compared with
conventional karyotyping, CMA is able to detect copy number variants
(CNVs) with much finer resolution and is not reliant on staining and
visual resolution limits. In 2010, a review of 33 published
studies—involving 21 698 patients with DD, congenital anomalies, or
autism—found the diagnostic yield of CMA to be 15% to 20% across all
studies, compared with 3% for the standard G-banded karyotype.9 In a group of 94 patients with no symptoms other than
ID, and no clear dysmorphic features, the diagnostic yield was 6.4%.8 According to the American Academy of Neurology
guideline in 2011, CMA testing was abnormal in approximately 7.8% of
patients with global DD or ID. The yield was higher (10.6%) in those with
syndromic features.8
Children with ASD who had co-morbid ID were more
likely to yield molecular diagnoses.10
Approximately 10% of patients with ASD exhibit a de-novo CNV, as detected
by CMA.11 Among ASD children
without syndromic features, only 6% received a molecular diagnosis.
In Hong Kong, two studies have investigated the use
of CMA in patients with DD, ID, ASD, or multiple congenital anomalies
(MCA). Chong et al12 found
clinically significant CMA results in 20 of 105 patients (19%). Tao et al13 found a diagnostic yield of 11%
for pathogenic or likely pathogenic results in 327 children, ages 1 month
to >20 years. Excluding patients with MCA, the diagnostic yield of CMA
for DD, ID, or ASD was approximately 4.2%.13
Chromosomal microarray has high clinical utility.
Firstly, it shortens the diagnostic odyssey and may avoid unnecessary
investigations, which reduces both individual and societal costs
associated with testing and medical care.8
14 Secondly, it may lead to a
clinically actionable recommendation. The prognostic information related
to diagnosis from CMA may alert other potential co-morbid conditions that
cannot be predicted on the basis of physical examination alone. In a
retrospective review of 1792 patients with DD, ID, ASD, or MCA who
underwent CMA testing, individuals with a positive diagnosis had a higher
rate of clinical actionable recommendations than those with an uncertain
result (54% vs 34%, P=0.01).15 In
Hong Kong, a detection rate of 8.6% was reported for clinically actionable
CNVs,13 which was comparable to the reported rates of 3.6% to 7% in
Western studies.15 16 17 Thirdly,
it allows estimation of recurrence risk and informed decisions regarding
reproductive options for the parents’ future pregnancies.
Children’s cognitive development at ≥5 years of age
is more stable if the level of ID is known. Children with DD undergo
assessment at CAS to facilitate their transition into primary school.
Since 2012, the Clinical Genetic Service (CGS) of the Department of Health
has provided CMA testing for DD, ID, or ASD. The presence of dysmorphic
features, early onset of DD, increased severity of DD, and family history
are common features that prompt a genetic referral. Collaboration between
CAS and CGS can potentially narrow the service gap for children with DD,
ID, or ASD by enabling early access to diagnostic genomic testing, thus
facilitating shorter waiting time for genetic and genomic investigation(s)
and a more client-friendly turnover time for results.
The aim of this study was to investigate the
genetic profile and diagnostic yield of CMA in children with moderate,
severe, and profound ID. The data obtained from this study are expected to
be useful in future service planning for children with DD or ID. The
diagnostic yield of CMA for children with more severe forms of ID is
suspected to be higher than the generally quoted figures of CMA (10%) for
investigation of ID. This study targeted children with more significant
ID, which is more likely to be related to an underlying genetic aetiology.
Methods
This cross-sectional territory-wide study recruited
children who attended CAS for developmental assessments, before Primary 1
entry, over a 12-month period from July 2016 to June 2017. All children
were at least 5 years of age. Inclusion criteria were children with
moderate, severe, or profound ID, with or without ASD. According to the
International Statistical Classification of Diseases and Related Health
Problems, tenth revision,18
moderate ID was defined as IQ 35 to 49; severe ID was defined as IQ 20 to
34; and profound IQ was defined as IQ <20. Exclusion criteria included
known causes of ID: (i) antenatal causes such as congenital brain
malformation or intrauterine infections; (ii) perinatal causes such as
prematurity (<34 weeks), birth asphyxia, or hypoxic ischaemic
encephalopathy; (iii) postnatal causes such as intracranial bleeding,
intracranial infection, or brain trauma; and (iv) other biological causes
such as inborn errors of metabolism, brain tumour, neuromuscular
disorders, neurodegenerative disorders, or cerebral palsy.
Unexplained ID in this study was defined as
children with no identifiable causes for ID who did not meet any of the
exclusion criteria. These children were non-syndromic and non-dysmorphic.
In addition, they had neither MCA nor family history of ID or ASD among
first- and second-degree relatives. The presence of MCA was defined as the
involvement of two or more organ systems.
Children were assessed by paediatricians with the
Griffiths Mental Developmental Scales,19
or by clinical psychologists with the Wechsler Preschool and Primary Scale
of Intelligence–Revised.20 The
Diagnostic and Statistical Manual of Mental Disorders, 4th edition,21 ASD diagnostic criteria were used for assessment of
ASD.
Genetic profile
For children with genetic diagnoses or who were
known clients of CGS, their medical files were retrieved from CAS and CGS
for review. Children with syndromic or dysmorphic features, MCA, or
significant family history, who had not been previously referred to CGS,
were referred for a formal genetic consultation before genetic or genomic
investigations were recommended by a clinical geneticist.
An expedited pathway was offered for children with
unexplained ID. Pre-genetic counselling was provided by a paediatrician at
CAS, followed by direct blood examination for CMA and Fragile X syndrome
(FGX) testing at CGS. Consultation with a geneticist was arranged if
either CMA or Fragile X testing yielded abnormal outcomes. Otherwise,
clients did not consult a geneticist for further counselling.
Chromosomal microarray testing and interpretation
For each patient, 3 mL of blood in
ethylenediaminetetraacetic acid was sent to the laboratory at CGS. All
samples were tested by PerkinElmer CGXTM v2 60K arrays designed
by Agilent SurePrint technology, in accordance with the manufacturer’s
instructions. The coverage of the array demonstrated an average resolution
of 140 kb across the genome, and ≤40 kb in regions of clinical relevance.
It evaluated >245 known genetic syndromes and >980 gene regions of
functional significance in human development. Data were analysed by
Genoglyphix software (Signature Genomics, Spokane [WA], United States).
Genomic coordinates were based on genome assembly hg19.
Detected CNVs were systematically evaluated for
clinical significance by comparison with information in the proprietary
Genoglyphix Chromosome Aberration Database (Signature Genomics), internal
laboratory database at CGS and the Department of Health, and public
databases (Database of Genomic Variants, International Standards for
Cytogenomic Arrays Consortium, and Database of Chromosomal Imbalance and
Phenotype in Humans using Ensembl Resources). Categorisation of CNVs was
based on available phenotypes and comparison of phenotypes with genes in
the region of copy gain or loss. This was performed by searching the
following databases: Online Mendelian Inheritance in Man, PubMed, RefSeq,
and the University of California Santa Cruz genome browser.22 Confirmatory fluorescence in situ hybridisation
(FISH), multiplex ligation-dependent probe amplification (MLPA), or
conventional karyotyping was performed as indicated. Parental testing was
offered to aid further interpretation and classification. Copy number
variants were classified as pathogenic, likely pathogenic, uncertain
clinical significance, or benign, in accordance with the 2011 American
College of Medical Genetics practice guidelines.23
Only pathogenic and likely pathogenic CNVs were regarded as clinically
significant.
Sample size calculation
The number of subjects to be recruited was
estimated based on the average number of children with moderate, severe,
and profound ID in the CAS database. In 2013 to 2015, the average number
of children with moderate ID or worse was approximately 270 children per
year. With the assumption that 60% of cases were unexplained,7 potential
cases eligible for CMA were estimated as 160 children per year. Literature
showed that the diagnostic yield of CMA was 10% in identifying abnormal
cases in similar settings.13 A 95%
confidence interval was desired, with a reliability (d) of 0.05, in
obtaining a diagnostic yield (ˆp) of 10% in this study. The sample size
needed was determined following a previously published method1:
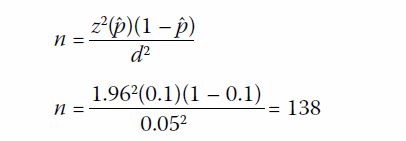
Hence, a target sample size of 138 was needed.24
Statistical analysis
The genetic profile of children in the study was
described. The diagnostic yield from CMA was calculated according to the
severity of ID. The Freeman-Halton test was used to test associations
between the severity of (a) ID and CMA and (b) dysmorphism and CMA
findings. The null hypothesis was that there was no association between
the severity of ID or dysmorphism and CMA findings. P<0.05 indicated an
association between the severity of ID or dysmorphism and CMA findings.
The Freeman-Halton test was conducted by using SAS/STAT 9.22.
Results
From July 2016 to June 2017, there were a total of
339 children diagnosed with more severe forms of ID: 241 (71%) children
had moderate ID, 49 (14.5%) had severe ID, and 49 (14.5%) had profound ID.
Eighty-three children were excluded for the following reasons: (1) they
met predefined exclusion criteria; (2) their family could not participate
due to geographical reasons (eg, family lived in China); (3) a language
barrier affected their understanding of study details (eg, the children or
their families spoke primarily Nepalese or Sri Lankan); or (4) their
parents could not be contacted for consent. A total of 31 children opted
not to participate in the study. In all, 225 (66.4%) of 339 children
participated in the study.
Among the 225 children, 116 (51.6%) had a co-morbid
diagnosis of ASD. Male (n=151) to female (n=74) ratio was 2:1. The age
ranged from 5 to 10 years old with a mean age of 6.6 years old. In all,
71.5% of children had moderate ID, 14.7% had severe ID, and 13.8% had
profound ID. Two hundred twenty-one (98%) children had Chinese parents.
There were two pairs of consanguineous parents: one Indian couple and one
Pakistani couple.
Genetic profile of children with intellectual
disability
As shown in Table 1, 68 (30.2%) children were diagnosed with a
genetic condition. The percentage of a positive genetic diagnosis
increased with the severity of ID. Chromosomal abnormalities comprised 76%
(n=52) of the total genetic diagnoses. The most common syndromic diagnosis
was Down syndrome (n=22). There were two cases of FGX. Three children had
chromosome 22 microdeletion syndromes—one exhibited the more common
chromosome 22q11.2 microdeletion syndrome (DiGeorge syndrome), whereas the
other two exhibited chromosome 22q13.3 deletion syndrome.
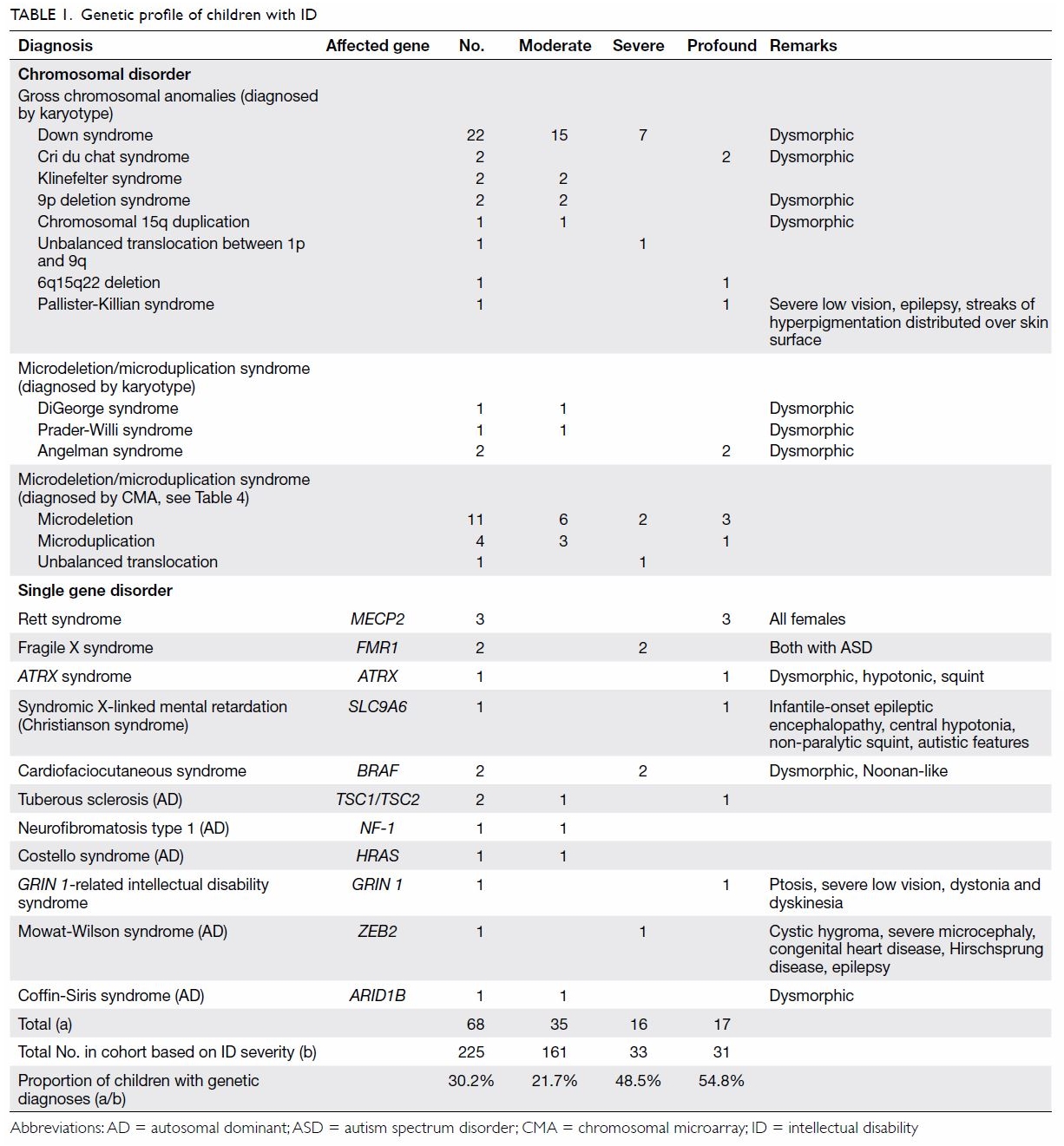
Table 1. Genetic profile of children with ID
Diagnostic yield of chromosomal microarray in children
with intellectual disability
Of the 225 participating children, 138 underwent
CMA testing; 53 (38%) children were referred to the Clinical Genetic
Service by the expedited referral pathway. Table 2 shows that 16 (11.6%) children demonstrated
clinically significant CNVs that explained their ID phenotype and 10
(7.2%) had variants of uncertain significance (VUS). The diagnostic yield
of CMA increased with severity of ID: it was 8.7% in moderate ID, 17.6% in
severe ID, and 23.5% in profound ID (P<0.05; Table 3). Among the 16 children with clinically
significant CNVs, 11 demonstrated copy number loss (deletion), four
demonstrated copy number gain (duplication), and one demonstrated an
unbalanced translocation between chromosome 7q and 20p (Table
4). One case of Angelman syndrome was detected by CMA and later
confirmed with MLPA. One case of Cri du chat syndrome was detected by CMA
and later confirmed with FISH. In total, 69% of pathogenic or likely
pathogenic CNVs were de novo. Ten children (7.2%) were incidentally
identified as carriers of disease: six were alpha thalassemia heterozygous
carriers, one was a heterozygous carrier of Joubert syndrome type 4, one
was a heterozygous carrier of autosomal recessive disease Joubert syndrome
and nephronophthisis, one was a heterozygous carrier of autosomal
recessive deafness affecting the OTOA gene, and one was a carrier
of Klinefelter syndrome.

Table 2. Chromosomal microarray diagnostic yields in unexplained ID
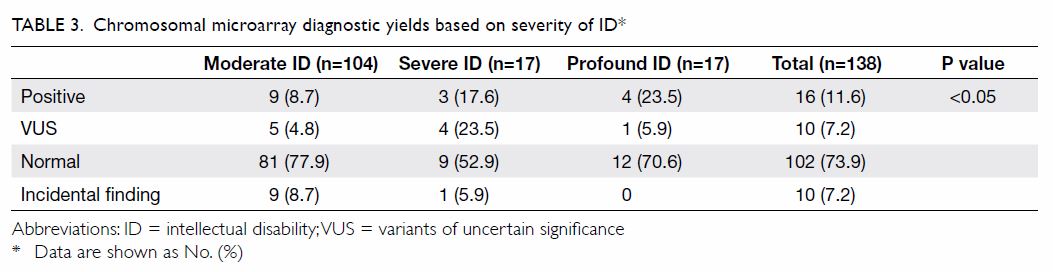
Table 3. Chromosomal microarray diagnostic yields based on severity of ID
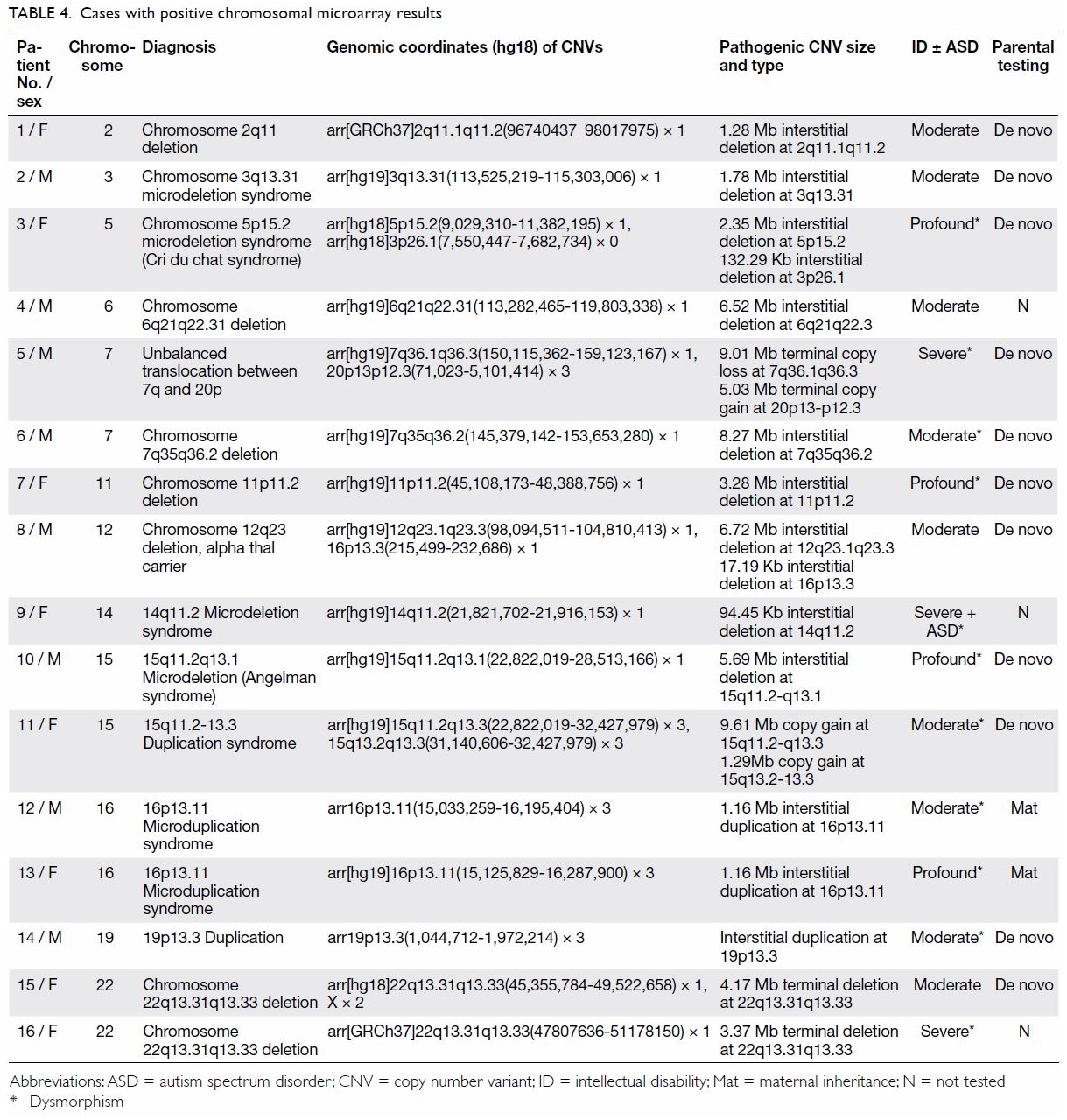
Table 4. Cases with positive chromosomal microarray results
Discussion
The overall diagnostic yield of CMA among children
with ID (11.6%) was consistent with studies performed in other regions of
the world. The diagnostic yield of CMA increased with severity of ID and
was much higher in children with dysmorphism (45.8% vs 4.4%, P<0.05).
Variants of uncertain significance are not
uncommon. In all, 7.2% of children in this cohort had VUS (Table
5). Regular follow-up and reassessment by a clinical geneticist is
necessary for these children, because VUS may eventually be re-classified
as pathogenic or benign as clinical and genomic data accumulate in the
literature.

Table 5. Cases with variants of uncertain significance
The paradigm shift in the medical genetic and
genomic field from the phenotype-first approach to the genotype-first
approach is revolutionary. Traditionally, a phenotype-first approach was
used to guide the investigation of possible genetic diagnoses, eg,
karyotyping for Down syndrome, or specific assays, such as FISH, for
DiGeorge syndrome. In the past decade, CMA has allowed more comprehensive
unbiased discovery of microdeletion and microduplication syndromes
throughout the human genome. Since the 1980s, it has been well-known that
certain chromosomal microdeletion and microduplication syndromes are
associated with recognisable forms of ID and DD. Classical examples
include 15q11-q13 deletion, associated with Prader-Willi and Angelman
syndromes, and 22q11.2 deletion, associated with DiGeorge syndrome
(velocardiofacial syndrome). Thus far, approximately 50 to 60 recurrent
microdeletion or duplication syndromes have been identified in children
with DD or ID.
Although CMA is robust, it cannot replace a formal
genetic consultation for children with clinically suspected genetic
conditions. As an example, in Prader-Willi syndrome, 70% to 75% of cases
can be detected by CMA, as they are due to a paternal 15q microdeletion
subtype; 20% to 25% of cases require a more specific methodology for
genetic confirmation. Therefore, clinical correlation and expert
assessment remain necessary.
Males are more susceptible to ID than females; more
than 100 X-linked genes are associated with ID.25
X-linked ID constitutes 5% to 10% of ID in males. One of the best-known
causative genes for ID is FMR1; mutations of FMR1 result
in FGX. The estimated incidence of FGX is approximately 1 in 4000 males
and 1 in 5000 to 1 in 8000 females (approximately 0.5% of cases of ID) in
Western countries. Peprah26
reported that the incidence of FGX in countries/regions with significant
Asian populations, such as Canada, Estonia, Japan, and Taiwan, was
significantly lower than in Western countries. In a study of 553 male
children between the ages of 6 months and 18 years, Chen et al27 estimated the prevalence of FGX in mainland China to
be approximately 0.93% among children with moderate to severe ID. Among
the 225 children in our cohort, only two were diagnosed with FGX. Both
exhibited ASD and severe ID. The typical physical characteristics of FGX,
such as narrow face, protruding ears, and macro-orchidism, are often less
obvious in early childhood; notably, they may become more prominent as the
child approaches adolescence. This lack of early physical characteristics
increases the diagnostic challenge for clinicians. Fragile X syndrome
testing, regarded as first-tier genetic testing for DD and ASD in many
international guidelines, has been a standard genetic investigation for ID
or ASD in Hong Kong for many years.
Incomplete penetrance of a genomic condition within
the same family is not uncommon. Notably, there were two such cases of
16p13.11 microduplication syndrome in this cohort. Patient 12 (Table
4) exhibited subtle dysmorphism comprising downslanting palpebral
fissures, prominent ears, and mild right ptosis. Left undescended testes
and umbilical hernia were operated in infancy. He exhibited global DD and
was later diagnosed with moderate ID. His mother and two sisters had an
identical chromosomal defect, but exhibited normal intelligence. Patient
13 demonstrated a more severe phenotype with hirsutism, bushy eyebrows,
frontal bossing, hearing loss, visual problems, and profound ID. His
mother and elder brother, both carrying the microduplication, exhibited
normal intelligence. There likely exist unknown environmental or genetic
modifiers to modulate susceptibility to ID caused by this
microduplication. Thus, relying on family history to determine whether ID
is hereditary can be misleading.
An important aspect with respect to obtaining a
genetic diagnosis is patient prognosis. The 16p13.11 duplication syndrome
is associated with an aortic root defect. In this study, the two affected
children and their affected family members were referred for monitoring by
echocardiogram. Similarly, Patient 2 exhibited 3q13.31 microdeletion
syndrome, which is associated with diabetes mellitus and deafness; this
patient was referred for audiological assessment and counselled on
lifestyle management to minimise the risk of diabetes. In this study,
three of 16 CMA-positive cases (18.8%) were clinically actionable.
Pre-test genetic counselling is as important as
post-test counselling. Coincidental findings of genetic changes that
either predict adult-onset conditions or reveal carrier status for
recessive or X-linked conditions are common. In the present cohort, 10
children were identified as carriers of genetic conditions, including one
child diagnosed with Klinefelter syndrome. He presented with moderate ID
and ASD. Chromosomal microarray identified a copy number gain of the
entire X chromosome. Klinefelter syndrome can be associated with learning
disabilities, as well as delayed speech and language development. While a
small, but significant, downward shift in mean overall IQ has been
reported, general cognitive abilities of patients with Klinefelter
syndrome are not typically in the ID range.28
An extra X chromosome may have contributed partially, but could not
entirely explain the severity of ID. The major implications are that
individuals with Klinefelter syndrome have a higher risk of endocrine
dysfunction, fertility problems, male breast cancer, and autoimmune
disease.
This study provided important information with
respect to service planning for children with ID in Hong Kong. It allowed
testing of an expedited referral mechanism between CAS and CGS, in which
cases with unexplained ID benefitted through a significant reduction of
waiting time for both pre-testing genetic counselling and investigation
turnover time. This study included 38% of the 138 children who were not
referred to CGS. The ideal future approach may be to extend the expedited
mechanism for children with early-onset significant DD. It can avoid
unnecessary investigations, thus lowering stress for both child and
parent; importantly, it may reduce societal costs.
There were several limitations in this study.
Firstly, a complete genetic profile of ID was not generated, as this
cohort excluded mild ID. Secondly, clients from minority cultural groups
in Hong Kong were underrepresented, because the language barrier affected
recruitment. More effort must be expended to ensure equal opportunities
for children from diverse cultural backgrounds. Thirdly, the duration of
the study was of insufficient length for commentary on trends regarding
the genetic profile of ID in Hong Kong.
Conclusion
The overall diagnostic yield (11.6%) of CMA is
compatible with other international cohorts. Chromosomal microarray yield
increases with the severity of ID. These data further support the use of
CMA as a first-tier investigation for children with significant
unexplained ID in Hong Kong.
Author contributions
All authors have made substantial contributions to
the concept or design of the study, acquisition of data, analysis or
interpretation of data, drafting of the article, and critical revision for
important intellectual content.
Acknowledgement
The authors would like to thank Mr Morris Wu of
Child Assessment Service, Department of Health for his advice on the
statistical analysis of the data.
Declaration
The authors have no conflicts of interest to
disclose. All authors had full access to the data, contributed to the
study, approved the final version for publication, and take responsibility
for its accuracy and integrity.
Funding/support
This research received no specific grant from any
funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Approval was obtained from the Ethics Committee of
the Department of Health, Hong Kong Special Administrative Region.
Informed consent was obtained from parents or legal guardians. Parents and
legal guardians were counselled about the indication for CMA, benefits and
limitations of test, methodology, reporting time, and possible outcomes
upon recruitment.
References
1. Leonard H, Wen X. The epidemiology of
mental retardation: challenges and opportunities in the new millennium.
Ment Retard Dev Disabil Res Rev 2002;8:117-34. Crossref
2. Social Data Collected via the General
Household Survey. Special Topics Report No 62. Persons with Disabilities
and Chronic Diseases. Hong Kong: Census and Statistics Department, Hong
Kong SAR Government; 2014.
3. American Association on Intellectual and
Developmental Disabilities. Intellectual Disability: Definition,
Classification, and Systems of Supports. 11th ed. Washington, DC: American
Association on Intellectual and Developmental Disabilities;2010: xvi, 259.
4. Moeschler JB, Shevell M, Committee on
Genetics. Comprehensive evaluation of the child with intellectual
disability or global developmental delays. Pediatrics 2014;134:e903-18. Crossref
5. Tang KM, Chen TY, Lau VW, Wu MM.
Clinical profile of young children with mental retardation and
developmental delay in Hong Kong. Hong Kong Med J 2008;14:97-102.
6. Willemsen MH, Kleefstra T. Making
headway with genetic diagnostics of intellectual disabilities. Clin Genet
2014;85:101-10. Crossref
7. Rauch A, Hoyer J, Guth S, et al.
Diagnostic yield of various genetic approaches in patients with
unexplained developmental delay or mental retardation. Am J Med Genet A
2006;140:2063-74. Crossref
8. Michelson DJ, Shevell MI, Sherr EH,
Moeschler JB, Gropman AL, Ashwal S. Evidence report: Genetic and metabolic
testing on children with global developmental delay: report of the Quality
Standards Subcommittee of the American Academy of Neurology and the
Practice Committee of the Child Neurology Society. Neurology
2011;77:1629-35. Crossref
9. Miller DT, Adam MP, Aradhya S, et al.
Consensus statement: chromosomal microarray is a first-tier clinical
diagnostic test for individuals with developmental disabilities or
congenital anomalies. Am J Hum Genet 2010;86:749-64. Crossref
10. Tammimies K, Marshall CR, Walker S, et
al. Molecular diagnostic yield of chromosomal microarray analysis and
whole-exome sequencing in children with autism spectrum disorder. JAMA
2015;314:895-903. Crossref
11. Sebat J, Lakshmi B, Malhotra D, et al.
Strong association of de novo copy number mutations with autism. Science
2007;316:445-9. Crossref
12. Chong WW, Lo IF, Lam ST, et al.
Performance of chromosomal microarray for patients with intellectual
disabilities/developmental delay, autism, and multiple congenital
anomalies in a Chinese cohort. Mol Cytogenet 2014;7:34. Crossref
13. Tao VQ, Chan KY, Chu YW, et al. The
clinical impact of chromosomal microarray on paediatric care in Hong Kong.
PLoS One 2014;9:e109629. Crossref
14. Tirosh E, Jaffe M. Global
developmental delay and mental retardation—a pediatric perspective. Dev
Disabil Res Rev 2011;17:85-92. Crossref
15. Coulter ME, Miller DT, Harris DJ, et
al. Chromosomal microarray testing influences medical management. Genet
Med 2011;13:770-6. Crossref
16. Ellison JW, Ravnan JB, Rosenfeld JA,
et al. Clinical utility of chromosomal microarray analysis. Pediatrics
2012;130:e1085-95. Crossref
17. Riggs ER, Wain KE, Riethmaier D, et
al. Chromosomal microarray impacts clinical management. Clin Genet
2014;85:147-53. Crossref
18. World Health Organization. The ICD-10
classification of mental and behavioural disorders: diagnostic criteria
for research: Geneva: World Health Organization; 1993.
19. Luiz D, Barnard A, Knosen N, Kotras N,
Faragher B, Burns LE. Griffiths Mental Development Scales–Extended
Revised. Two to Eight Years. Administration Manual. Oxford, UK: Hogrefe;
2006.
20. Wechsler D. Wechsler Preschool and
Primary Scale of Intelligence–Revised. WPPSI-R: Psychological Corporation;
1989.
21. Diagnostic and Statistical Manual of
Mental Disorders: DSM-IV. Washington, DC: American Psychiatric
Association;1994: 535.
22. Kan AS, Lau ET, Tang WF, et al.
Whole-genome array CGH evaluation for replacing prenatal karyotyping in
Hong Kong. PLoS One 2014;9:e87988. Crossref
23. Kearney HM, Thorland EC, Brown KK, et
al. American College of Medical Genetics standards and guidelines for
interpretation and reporting of postnatal constitutional copy number
variants. Genet Med 2011;13:680-5. Crossref
24. Boada R, Janusz J, Hutaff-Lee C,
Tartaglia N. The cognitive phenotype in Klinefelter syndrome: a review of
the literature including genetic and hormonal factors. Dev Disabil Res Rev
2009;15:284-94. Crossref
25. Lubs HA, Stevenson RE, Schwartz CE.
Fragile X and Xlinked intellectual disability: four decades of discovery.
Am J Hum Genet 2012;90:579-90. Crossref
26. Peprah E. Fragile X syndrome: the FMR1
CGG repeat distribution among world populations. Ann Hum Genet
2012;76:178-91. Crossref
27. Chen X, Wang J, Xie H, et al. Fragile
X syndrome screening in Chinese children with unknown intellectual
developmental disorder. BMC Pediatr 2015;15:77. Crossref
28. Daniel WW. Biostatistics: A Foundation
for Analysis in the Health Sciences. 9th ed. Hoboken, NJ: J Wiley &
Sons; 2009.