DOI: 10.12809/hkmj166048
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Chronic mucocutaneous candidiasis—more than just skin
deep
Philip H Li, MB, BS, MRCP(UK)1; Pamela
PW Lee, MD, FHKAM (Paediatrics)2; SL Fung, MB, BS, FHKAM
(Medicine)3; CS Lau, MD, FRCP (Edin, Glasg, Lond)1;
YL Lau, MD, FRCPCH (UK)2
1 Department of Medicine, Queen
Mary Hospital, Pokfulam, Hong Kong
2 Department Paediatrics and
Adolescent Medicine, Queen Mary Hospital, Pokfulam, Hong Kong
3 Tuberculosis and Chest Unit,
Grantham Hospital, Wong Chuk Hang, Hong Kong
Corresponding author: Dr Philip H Li (philipli@connect.hku.hk)
 Full
paper in PDF
Full
paper in PDF
Case presentation
A 24-year-old Chinese man was referred to the Chest
Unit of Grantham Hospital, Hong Kong in June 2016 for management of
bronchiectasis. The patient had experienced his first episode of lower
respiratory tract infection (LRTI) at age 17 years, which required
hospital admission for intravenous antibiotics. He had since developed
four to five episodes of LRTI annually and subsequently developed
bronchiectasis with daily copious sputum and occasional haemoptysis.
Review of the patient’s records revealed a history
of autoimmune hypothyroidism since age 12 years, which was based on
thyroid scintigraphy and high titres of antithyroid autoantibodies and
required thyroxine replacement. At age 21 years, after an incidental
finding of iron deficiency anaemia, the patient underwent upper endoscopy,
which revealed oesophageal candidiasis (Fig 1). The patient had recurrent and chronic oral
ulcers, and excisional biopsy with Grocott staining displayed fungal
spores and pseudohyphae consistent with candidiasis.
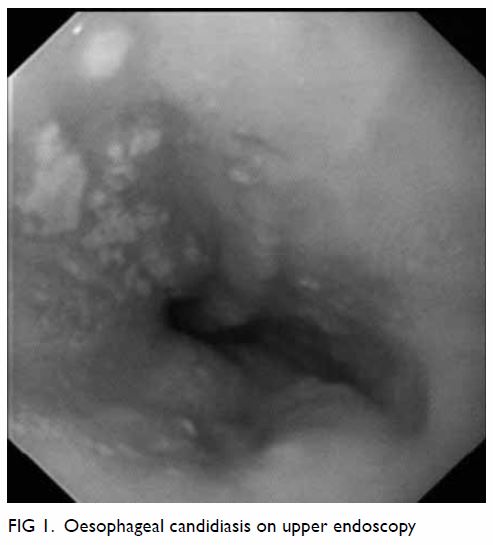
Figure 1. Oesophageal candidiasis on upper endoscopy
Further questioning of the patient revealed
frequent tinea infections and orogenital candidiasis since early
childhood. The patient had had childhood chickenpox with two further
episodes of zoster reactivation during his teens. There were no features
suggestive of atopy nor any history of recurrent abscesses. The patient
had difficulty attending his frequent medical appointments and often
required sick leave from his work as a technician. He was a never smoker
and social drinker. The patient was born and raised in Hong Kong, and had
received all routine vaccinations without problems. The family history was
unremarkable with no history of consanguinity. The patient lived with his
parents and younger brother, none of whom had candidiasis.
High-resolution computed tomography revealed
bilateral lower lobe bronchiectasis (Fig 2) and bronchoscopy again showed extensive
candidiasis with no airway abnormalities. Pulmonary function test results
and serum alpha-1-antitrypsin level were normal. Sputum cultures were
negative for acid-fast bacilli and yeasts, and aerobic culture yielded
commensals only. The patient’s complete blood counts were grossly normal
apart from a persistently low absolute lymphocyte count (0.5-0.7 × 109
/L). Immunoglobulin (Ig) levels (IgG/IgM/IgA/IgE) were all within normal
limits. Screening for diabetes mellitus and testing for human
immunodeficiency virus (HIV) was negative. Complement levels and the
dihydrorhodamine assay were normal. A serology panel also found positive
anti-nuclear antibodies with a titre of 1:320, in addition to previously
documented thyroid autoantibodies. Repeated lymphocyte subsets showed
persistent panlymphocytopenia (CD19:43/μL[n:91-452],
CD3:491/μL[n:938-2311], CD4:206/μL[n:437-1226], CD8:263/μL[n:322-1104] and
CD16/56:115/μL[n:177-1059]), with impaired lymphocyte proliferation after
phytohaemagglutinin and pokeweed mitogen stimulation.
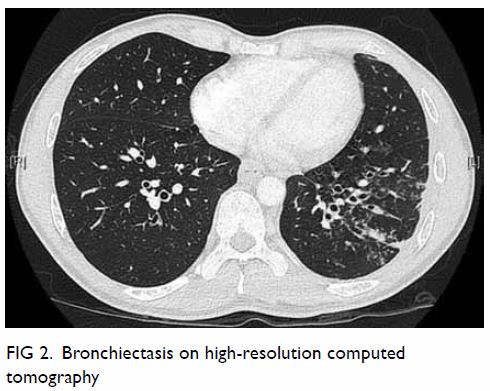
Figure 2. Bronchiectasis on high-resolution computed tomography
In view of his chronic mucocutaneous candidiasis
(CMC), autoimmunity and combined immunodeficiency, the patient was
referred to us for further evaluation. An interleukin (IL)-17–related
defect was suspected and sequencing of the signal transducer and activator
of transcription (STAT) 1 gene was performed. A previously reported
gain-of-function (GOF) mutation of p.G384D in the DNA-binding domain was
identified.1 The patient was
counselled and long-term itraconazole and co-trimoxazole prophylaxis
treatment was started. Measurement of pneumococcal antibody responses were
scheduled. The patient continues to receive multidisciplinary care between
immunology, pulmonology, and internal medicine after a unifying diagnosis
for his wide spectrum of disease manifestations was made.
Discussion
That primary immunodeficiencies (PIDs) only affect
children is a common misconception. Diagnoses are also made in adulthood
due to genuine adult-onset types, as in our case, or delayed disease
recognition. We describe the first reported adult case of STAT1-GOF
mutation in our locality. The patient had a typical history of CMC,
complicated by recurrent infections, bronchiectasis, and autoimmune
hypothyroidism. The patient also had panlymphocytopenia, which is reported
in 20% to 30% of STAT1-GOF patients and associated with LRTIs.2 The first possible indication of STAT1-GOF mutation was
the autoimmune hypothyroidism in the context of CMC.
Typically, CMC is characterised by
persistent/recurrent non-invasive Candida infections of the skin, nails,
and mucous membranes. Candidiasis is usually an opportunistic infection
and associated with immunosuppression acquired through diabetes mellitus
or HIV, or use of chemotherapy, glucocorticoids, or antibiotics.
Candidiasis is also associated with a variety of PIDs, usually with an
underlying IL-17 pathway defect. Examples include hyper-IgE syndromes,
autoimmune polyendocrinopathy syndrome type 1, CARD9 deficiency, in
addition to IL-17F and IL-17RA deficiencies.3
Since 2011, STAT1-GOF mutations have been
discovered that cause autosomal dominant familial CMC. These mutations are
now established as the most common genetic cause of inherited CMC. Such
STAT1-GOF mutations have also been reported in paediatric patients in Hong
Kong with CMC and penicilliosis.4
These mutations impair STAT1 dephosphorylation and enhance the production
of STAT1-dependent cytokines (interferon-α/β, interferon-γ and IL-27). In
turn, this likely represses STAT3-dependent gene transcription and impairs
the development of IL-17–producing T cells, although the exact mechanisms
remain unclear.5
A recent analysis of the clinical manifestations in
274 individuals with 76 different STAT1-GOF mutations revealed an
extremely broad disease phenotype.2
In addition to CMC, patients were also prone to other fungal, bacterial
(usually LRTIs), and viral infections. Results of immunological
investigations were variable, but abnormalities were significantly
associated with increased infections. More than a third of patients had
autoimmune/inflammatory disorders, most commonly hypothyroidism, and 21%
of patients had bronchiectasis.2
Most patients with autoimmunity had positive autoantibodies. Alarmingly,
these patients are more likely to develop other autoimmune disorders,
cerebral/abdominal aneurysms, and squamous cell carcinomas (likely
secondary to chronic mucocutaneous inflammation).
The outcome for patients with STAT1-GOF mutations
remains poor, with most deaths resulting from infection, aneurysms, or
malignancies. Long-term (as opposed to intermittent) systemic antifungal
therapy remains the mainstay of treatment, aiming to reduce the
development of antifungal resistance and malignancy. Additional antibiotic
prophylaxis and intravenous Ig should be considered for patients with
recurrent infections. In our patient, intravenous Ig would have been
indicated if his response to pneumococcal vaccination was suboptimal in
the context of bronchiectasis. Furthermore, autoimmune hypothyroidism is
associated with cerebral aneurysms and baseline magnetic resonance
angiography may be indicated. Lastly, any ear, nose, and throat or
gastrointestinal symptoms should alert the physician to a need for regular
monitoring of such malignancies with biopsy. Experimental therapies such
as colony-stimulating factors and ruxolitinib, a Janus kinase 1/2
inhibitor, have been reported to successfully improve CMC and will merit a
trial if his CMC worsens with time.6
For other PIDs, the main goals are to reduce the
incidence of infections and to prevent development of complications; with
the ultimate aim of cure. Improved awareness and further development of
adult clinical immunology in Hong Kong is required, so that adult patients
with PID receive more timely and comprehensive care.
Author contributions
All authors have made substantial contributions to
the concept; acquisition of data; interpretation of data; drafting of the
article; and critical revision for important intellectual content.
Declaration
All authors have disclosed no conflicts of
interest. All authors had full access to the data, contributed to the
study, approved the final version for publication, and take responsibility
for its accuracy and integrity.
References
1. Yamazaki Y, Yamada M, Kawai T, et al.
Two novel gain-of-function mutations of STAT1 responsible for chronic
mucocutaneous candidiasis disease: impaired production of IL-17A and
IL-22, and the presence of anti-IL-17F autoantibody. J Immunol
2014;193:4880-7. Crossref
2. Toubiana J, Okada S, Hiller J, et al.
Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly
broad clinical phenotype. Blood 2016;127:3154-64. Crossref
3. Puel A, Cypowyj S, Maródi L, Abel L,
Picard C, Casanova JL. Inborn errors of human IL-17 immunity underlie
chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol
2012;12:616-22. Crossref
4. Lee PP, Mao H, Yang W, et al. Penicillium
marneffei infection and impaired IFN-γ immunity in humans with
autosomal-dominant gain-of-phosphorylation STAT1 mutations. J Allergy Clin
Immunol 2014;133:894-6.e5. Crossref
5. Zheng J, van de Veerdonk FL, Crossland
KL, et al. Gain-of- function STAT1 mutations impair STAT3 activity in
patients with chronic mucocutaneous candidiasis (CMC). Eur J Immunol
2015;45:2834-46. Crossref
6. van de Veerdonk FL, Netea MG. Treatment
options for chronic mucocutaneous candidiasis. J Infect 2016;72
Suppl:S56-60. Crossref