Hong
Kong Med J 2018 Aug;24(4):340–9 | Epub 2 Mar 2018
DOI: 10.12809/hkmj176870
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Genetic basis of channelopathies and cardiomyopathies
in Hong Kong Chinese patients: a 10-year regional laboratory experience
Chloe M Mak1; Sammy PL Chen2;
NS Mok3; WK Siu2; Hencher HC Lee2; CK
Ching2; PT Tsui3; NC Fong4; YP Yuen2;
WT Poon2; CY Law2; YK Chong2; YW Chan2;
TC Yung5; Katherine YY Fan6; CW Lam7
1 Chemical Pathology Laboratory, Kowloon
West Cluster Laboratory Genetic Service, Department of Pathology, Princess
Margaret Hospital, Laichikok, Hong Kong
2 Department of Pathology, Princess
Margaret Hospital, Laichikok, Hong Kong
3 Department of Medicine, Princess
Margaret Hospital, Laichikok, Hong Kong
4 Department of Paediatrics and
Adolescent Medicine, Princess Margaret Hospital, Laichikok, Hong Kong
5 Department of Paediatric Cardiology,
Queen Mary Hospital, Pokfulam, Hong Kong
6 Department of Cardiac Medicine,
Grantham Hospital, Wong Chuk Hang, Hong Kong
7 Department of Pathology, The
University of Hong Kong, Pokfulam, Hong Kong
Corresponding author: Dr Chloe M Mak (makm@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Introduction: Hereditary
channelopathies and cardiomyopathies are potentially lethal and are
clinically and genetically heterogeneous, involving at least 90 genes.
Genetic testing can provide an accurate diagnosis, guide treatment, and
enable cascade screening. The genetic basis among the Hong Kong Chinese
population is largely unknown. We aimed to report on 28 unrelated
patients with positive genetic findings detected from January 2006 to
December 2015.
Methods: Sanger sequencing was
performed for 28 unrelated patients with a clinical diagnosis of
channelopathies or cardiomyopathies, testing for the following genes: KCNQ1,
KCNH2, KCNE1, KCNE2, and SCN5A, for long
QT syndrome; SCN5A for Brugada syndrome; RYR2 for
catecholaminergic polymorphic ventricular tachycardia; MYH7 and
MYBPC3 for hypertrophic cardiomyopathy; LMNA for dilated
cardiomyopathy; and PKP2 and DSP for arrhythmogenic
right ventricular dysplasia/cardiomyopathy.
Results: The study included 17 male and
11 female patients; their mean age at diagnosis was 39 years (range, 1-80
years). The major clinical presentations included syncope, palpitations,
and abnormal electrocardiography findings. A family history was present
in 13 (46%) patients. There were 26 different heterozygous mutations
detected, of which six were novel—two in SCN5A
(NM_198056.2:c.429del and c.2024-11T>A), two in MYBPC3
(NM_000256.3:c.906-22G>A and c.2105_2106del), and two in LMNA
(NM_170707.3:c.73C>A and c.1209_1213dup).
Conclusions: We characterised the genetic heterogeneity in channelopathies and cardiomyopathies among Hong Kong Chinese patients in a 10-year case
series. Correct interpretation of genetic findings is difficult and
requires expertise and experience. Caution regarding issues of
non-penetrance, variable expressivity, phenotype-genotype correlation,
susceptibility risk, and digenic inheritance is necessary
for genetic counselling and cascade screening.
New knowledge added by this study
- We characterised the genetic heterogeneity in channelopathies and cardiomyopathies among Hong Kong Chinese patients and described 26 mutations with six novel variants.
- This is the first case series of cardiac genetics in Hong Kong.
- This study provides genetic information for variant interpretation and insight into the clinical application of genetic testing for channelopathies and cardiomyopathies.
Introduction
Cardiac genetics is evolving rapidly and many new
insights have recently been achieved. Genetic causes are found in various
potentially lethal channelopathies and cardiomyopathies including long and
short QT syndrome (LQTS and SQTS), Brugada syndrome, catecholaminergic
polymorphic ventricular tachycardia (CPVT), hypertrophic cardiomyopathy
(HCM), dilated cardiomyopathy (DCM), arrhythmogenic right ventricular
dysplasia/cardiomyopathy (ARVD/C), Barth syndrome, and left ventricular
non-compaction.1 Knowledge of
genetics deepens the understanding of pathophysiology and remarkably
changes the diagnosis, treatment, and genetic counselling for recurrence
risk and family planning. This group is highly genetically heterogeneous (Table 12).
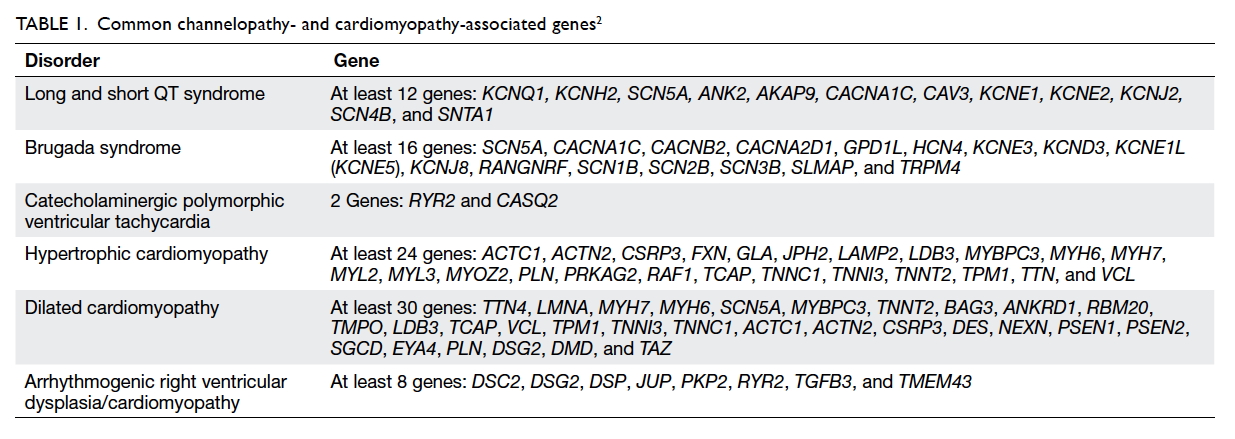
The genetic basis of inherited cardiac diseases in
the Hong Kong Chinese population is largely unknown. The Princess Margaret
Hospital provides a comprehensive cardiac genetic service. We conducted
this study to review the clinical and genetic findings of 28 unrelated
positive cases encountered between January 2006 and December 2015.
Methods
Diagnosis of the cardiac conditions was based on
clinical assessments by a cardiologist and practice guidelines.3 4 5 The patients were referred by cardiologists from
various public hospitals for genetic analysis. Only patients with positive
genetic findings are reported in this study. There were seven patients
with LQTS, two with Brugada syndrome, two with CPVT, nine with HCM, four
with DCM, and four with ARVD/C. Local ethics board approval was obtained.
Peripheral blood samples were collected from the proband after informed
consent was obtained. Genomic DNA was extracted using a QIAamp Blood Kit
(Qiagen, Hilden, Germany). The coding exons and the flanking introns (10
bp) of each gene were amplified by polymerase chain reaction. The primer
sequences and protocol are available on request. Sanger sequencing was
performed in the following order and stopped once a positive finding was
detected: KCNQ1, KCNH2, KCNE1, KCNE2, and
SCN5A for LQTS; SCN5A for Brugada syndrome; RYR2
for CPVT; MYH7 and MYBPC3 for HCM; LMNA for DCM;
and PKP2 and DSP for ARVD/C. The order was based on
prevalence according to the literature and local experience. All coding
exons were amplified for each gene except selected exons 3, 8, 14, 45, 46,
47, 49, 88, 89, 90, 93, 96, 97, 100, 101, and 103 for RYR2.6 The GenBank accession numbers are shown in Table
2. The pathogenicity of novel missense variants was analysed by
Alamut Visual (Interactive Biosoftware, Rouen, France) with Polymorphism
Phenotyping v2 (PolyPhen-2), Sorting Intolerant from Tolerant (SIFT),
MutationTaster, and Assessing Pathogenicity Probability in Arrhythmia by
Integrating Statistical Evidence (APPRAISE,
https://cardiodb.org/APPRAISE/) and that of novel splicing variants by
Splice Site Finder-like, MaxEntScan, NNSPLIC, GeneSplicer, and Human
Splicing Finder, wherever appropriate. Splicing variants were considered
to be damaging if there was a >10% lower score when compared with the
wild-type prediction. Allele frequencies among populations were referred
to the Exome Aggregation Consortium (ExAC;
http://exac.broadinstitute.org/).

Table 2. Targeted genes in Sanger sequencing analysis
Results
During the 10-year study period more than 90 patients with
channelopathies or cardiomyopathies were referred for genetic analysis.
Among them, 28 unrelated patients had positive genetic results, comprising
17 males and 11 females. Their mean age at diagnosis was 39 years (range,
1-80 years). The major clinical presentations included syncope,
palpitations, and abnormal electrocardiography (ECG) findings. Four
patients were asymptomatic and were diagnosed following an incidental
abnormal finding related to other medical issues. A family history was
present in only 13 (46%) patients. All detected mutations were
heterozygous, and 26 different heterozygous mutations were detected. These
encompassed 11 missense, two nonsense, and five splicing mutations, as
well as eight small insertions and deletions. There were six novel
mutations—two in SCN5A (NM_198056.2:c.429del and c.2024-11T>A),
two in MYBPC3 (NM_000256.3:c.906-22G>A and c.2105_2106del), and
two in LMNA (NM_170707.3:c.73C>A and c.1209_1213dup) [Table
3]. All were considered pathogenic or likely pathogenic according to
the Practice Guidelines for the Evaluation of Pathogenicity and the
Reporting of Sequence Variants in Clinical Molecular Genetics by the
Association for Clinical Genetic Science.7
Further clinical details and genotypes are shown in Table
3.
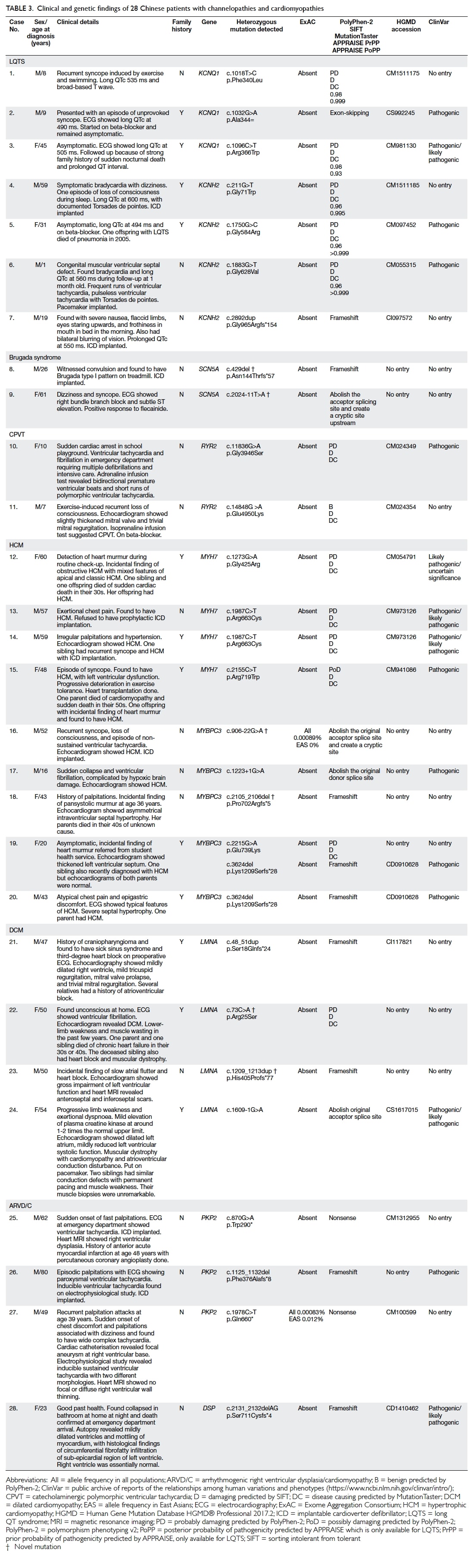
Table 3. Clinical and genetic findings of 28 Chinese patients with channelopathies and cardiomyopathies
There were seven patients with LQTS, two with
Brugada syndrome, two with CPVT, nine with HCM, four with DCM, and four
with ARVD/C. Three patients with LQTS had mutations in KCNQ1
(cases 1-3) and four had mutations in KCNH2 (cases 4-7). Two
patients (cases 8 and 9) with Brugada syndrome had mutations in SCN5A,
including two novel mutations. Two patients (cases 10 and 11) with CPVT
had mutations in RYR2. Four patients with HCM (cases 12-15) had MYH7
mutations and five (cases 16-20) had MYBPC3 mutations, including
two novel mutations. Four patients with DCM (cases 21-24) had LMNA
mutations, including two novel mutations. Finally, three patients with
ARVD/C had PKP2 mutations (cases 25-27) and one had a DSP
mutation (case 28).
Discussion
This is the first report of a cardiac genetic case
series among Hong Kong Chinese patients with channelopathies and
cardiomyopathies. A total of 28 patients are reported, and 26 different
mutations and six novel mutations have been identified. Wide genetic
diversity is observed, with no common mutation found. Hereditary
channelopathies and cardiomyopathies are mainly inherited in an autosomal
dominant manner. Mutations can be either inherited or de novo. Risk to
proband sibling(s) and first-degree relatives depends on the genetic
status of the parents. Offspring of the proband have a 50% risk of
inheriting the mutation. Siblings of the proband have the same risk if the
mutation is transmitted from either parent. Patients carrying a mutation
of these sudden arrhythmia death syndromes show incomplete penetrance. In
general, a mutation carrier will show symptoms/signs in 80% of those with
CPVT, 20% to 50% of ARVD/C patients, 18% to 63% of LQTS patients, 80% to
94% of SQTS patients, and 80% of patients with Brugada syndrome who have
abnormal ECG findings when challenged with a sodium channel blocker.8 No exact figure is available for HCM. The data could be
more specific if a particular mutation was considered alongside clinical
findings and family history. Pre-symptomatic testing of at-risk family
members cannot be used to predict age of onset, severity, type of
symptoms, or rate of progression. Detailed clinical, ECG, and genetic
characterisation of affected and unaffected family members is helpful.
Long QT syndrome
Long QT syndrome is genetically heterogeneous, with
at least 12 genes involved. Mutations in the four genes, KCNQ1, KCNH2,
KCNE1, and KCNE2, are detected in 46%, 38%, 2%, and 1% of
affected patients, respectively.8 A
small proportion of patients (3%) have double heterozygous mutations in
more than one disease loci.9
Specific arrhythmogenic triggers are associated with a particular subtype,
such as exertion, swimming, and near-drowning for LQT1; auditory triggers
and cardiac events occurring in the postpartum period for LQT2; and
cardiac events during sleep or at rest for LQT3. Three patients had KCNQ1
mutations. Case 1 had recurrent syncope induced by exercise and swimming,
but genetic testing confirmed LQTS type 1. Other patients had no specific
provoking factor. LQTS type 2 caused by KCNH2 mutations accounts
for about 38% of all LQTS.8 Four patients (cases 4-7) carried KCNH2
mutations and two (cases 4 and 6) presented with Torsades de pointes and
one (case 7) had survived cardiac arrest requiring an implantable
cardioverter defibrillator. Case 6 was the youngest patient, presenting at age 1 year. Genotype-guided treatment in LQTS is recommended and
LQT1 responds best to beta-blockers.10
11
Brugada syndrome
Brugada syndrome is characterised by cardiac
conduction abnormalities (ST-segment abnormalities in leads V1-V3 on ECG
and a high risk for ventricular arrhythmias) that can result in sudden
death. The Shanghai Score System has been recently published for the
diagnosis of Brugada syndrome.12 13 The prevalence of Brugada
syndrome or its characteristic ECG pattern is reportedly higher among
Asians, such as Japanese (0.14%-1.22%).14
15 16
17
Brugada syndrome is genetically heterogeneous and
can be attributed to defects in at least 23 genes at the time of
reporting.8 Mutations in SCN5A
are detected in 11% to 14% of affected individuals in Japan and <10% in
Taiwan where mutations in CACNA1C account for 1% to 7%.18 Approximately 65% to 70% of patients remain
genetically undiagnosed. Expressivity is variable and penetrance is
incomplete and low.
Conventionally, Brugada syndrome has been described
as a monogenic disease that has autosomal dominant inheritance with
incomplete penetrance; it is caused by rare genetic variants with a large
effect size. Most individuals diagnosed with Brugada syndrome have an
affected parent. The proportion of cases caused by a de-novo mutation is
approximately 1%. Recent studies indicate that genetic inheritance is
likely more complex, and models of an oligogenic disorder or
susceptibility risk/genetic predisposition have been suggested.19 20 21 22
Among the two patients in this series, none had a
positive family history. Symptoms were more non-specific, such as
palpitation and syncope. It is noteworthy that convulsion can be a
presentation of channelopathies (case 8). Clinical suspicion should be
higher with more specific investigations, such as exercise-stress ECG and
flecainide challenge tests, are required in order to reveal the real
culprit. Sudden cardiac death can be the first presenting symptom in
Brugada syndrome.
Two novel mutations are described in SCN5A:
c.429del and c.2024-11T>A. The former is predicted to cause a
frameshift and premature protein truncation. The latter is predicted to
abolish the acceptor splice site and create a cryptic site upstream. At
the time of reporting, both are absent from controls in the Exome
Sequencing Project, 1000 Genomes Project, and ExAC. SCN5A
mutations can cause either LQTS or Brugada syndrome.
Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular
tachycardia can present with syncope and sudden death during physical
exertion or emotion, due to catecholamine-induced bidirectional
ventricular tachycardia, polymorphic ventricular tachycardia or
ventricular fibrillation. The reported mean age of onset is between 7 and
12 years.8 Exercise stress testing
or an adrenaline provocation test may induce ventricular arrhythmia and
enable a clinical diagnosis. About half of these cases are related to a
dominantly inherited RYR2 gene mutation, with a small proportion
(1%-2%) related to recessively inherited CASQ2 gene mutations. RYR2
is a large gene with 105 exons. Tier testing has been proposed by
Medeiros-Domingo et al.6 First-tier
RYR2 genetic testing of the 16 selected exons allows
identification of about 65% of CPVT cases. There were two paediatric CPVT
patients (cases 10 and 11) in our series, with two known disease-causing
mutations detected, namely NM_001035.2(RYR2):c.11836G>A
(p.Gly3946Ser)23 24 25 26 and c.14848G>A (p.Glu4950Lys).23 24 Both
mutations were detected in first-tier screening.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy is the most prevalent
hereditary cardiac disease, causing about one third of sudden cardiac
deaths in young athletes. Its prevalence in China is approximately 1 in
1250.27 The clinical
manifestations are markedly variable, ranging from asymptomatic to sudden
cardiac death. Genetic testing provides an accurate diagnosis in the
probands and enables screening of asymptomatic family members. Although
the genetic background of HCM is heterogeneous, involving at least 30
genes, MYH7 and MYBPC3 are the most common and each
accounts for approximately 40%.8
Nine patients with HCM are reported here: four had
known MYH7 mutations and five had MYBPC3 mutations,
including two novel mutations. NM_000256.3:c.906-22G>A was detected in
case 16 and was a novel variant. Neither population frequency nor known
pathogenicity have been reported. In-silico analysis showed creation of a
novel acceptor site and insertion of 20 nucleotides into exon 10. This
conceivably would lead to a frameshift and premature protein termination.
Exon 10 of MYBPC3 is a microexon in which the stability of its
original splicing site is easily disrupted by intronic variants. A similar
mutation has been reported as c.906-36G>A.28
Nonetheless, cDNA analysis was not performed. NM_000256.3(MYBPC3):c.1223+1G>A
at the critical canonical +1 splice site is also novel. In addition, other
known disease-causing splicing mutations affecting the same nucleotide
have been reported.26 29 30 Case 19
had two variants detected in MYBPC3 (c.2215G>A and c.3624del).
The small deletion c.3624del is a mutation known to cause HCM in the
Chinese population31 and predicted
to cause a frameshift and premature termination of the protein. The
missense variant c.2215G>A is as yet unreported and is predicted by
in-silico analyses to cause an amino acid change from glutamate to lysine
at codon 739 and probably damage. At the time of reporting, the variant is
absent from controls in the Exome Sequencing Project, 1000 Genomes
Project, and ExAC databases. This variant is considered to have uncertain
significance. The mother of the patient in case 19 was available for
testing. She was 48 years old at the time of genetic testing,
asymptomatic, and heterozygous for c.3624del only. Hence, the two variants
c.2215G>A and c.3624del of MYBPC3 were in-trans in the patient
and elder brother of the patient in case 19. Both had a more severe form
of HCM, with a younger onset.
Dilated cardiomyopathy
Familial DCM is a group of genetically
heterogeneous disorders. Laminopathy can manifest as several allelic
disorders affecting muscle, nerve, adipose, and vascular tissues; one of
them is cardiomyopathy, dilated 1A. We identified four patients with DCM,
two of whom also had proximal muscle weakness. Two novel mutations in LMNA
were detected (c.73C>A and c.1209_1213dup). NM_005572.3(LMNA):c.73C>A
is a novel variant that is predicted to be deleterious by SIFT, probably
causing damage according to PolyPhen-2 and disease-causing according to
MutationTaster. Other missense mutations have been reported in the same
amino acid codon.32 33 34
NM_005572.3(LMNA):c.1609-1G>A is predicted to significantly
affect splicing by in-silico analysis. At the time of reporting, all
variants are absent from controls in the Exome Sequencing Project, 1000
Genomes Project, and ExAC. In case 22 with NM_005572.3(LMNA):c.73C>A,
one of the parents died of chronic heart failure in the fourth decade of
life, and one sibling died of heart block and chronic heart failure with a
diagnosis of muscular dystrophy at age 38 years. Nonetheless, there was no
sample left for genotyping.
Arrhythmogenic right ventricular
dysplasia/cardiomyopathy
Arrhythmogenic right ventricular
dysplasia/cardiomyopathy is associated with fibrofatty replacement of
cardiomyocytes, ventricular tachyarrhythmias, and sudden cardiac death.
Although the right ventricle is primarily affected in this condition,
left-dominant arrhythmogenic cardiomyopathy has also been described, and
mutations have been identified in DSP as well as in other genes.35 Four patients are reported here,
with three having mutations in PKP2 and one in DSP.
Interestingly, the patient in case 26 presented at age 80 years with
episodic palpitations. His ECG results showed paroxysmal ventricular
tachycardia. He had a deletion in PKP2, c.1125_1132del
(p.Phe376Alafs*8), resulting in a truncated incomplete protein product.
Age of onset in patients with PKP2 mutations is older than that of
the patient with DSP mutation. The latter patient (case 28) died
at age 23 years, with sudden collapse as the first presentation.
Primary arrhythmogenic disorders including
LQTS/SQTS, CPVT, Brugada syndrome, and cardiomyopathies account for about
one third of sudden cardiac deaths in the young.36
Identification of a pathogenic variant can solve the diagnostic mystery,
provide relief to the family, and enable family screening and counselling
for other at-risk family members. In some developed countries, molecular
autopsy is an essential part of a formal forensic investigation in
unexplained sudden death.37 We
support the implementation of molecular autopsy in routine autopsy
investigation of sudden cardiac death victims. Our group has conducted the
first local prospective study to determine the prevalence and types of
sudden arrhythmia death syndrome underlying sudden cardiac death among
local young victims through clinical and molecular autopsy of sudden
cardiac death victims and clinical and genetic evaluation of their
first-degree relatives (http://www.sadshk.org/en/medical_research.php).
Such data can serve as the groundwork for the feasibility of
implementation of such investigations in Hong Kong.
Genetic tests for cardiac conditions can aid
diagnosis and guide treatment. Nonetheless, there are limitations that
complicate the translational use of genetic results in patient care, such
as incomplete penetrance, variable expressivity, and findings of variants
of uncertain significance. In addition, since the genetic heterogeneity is
large among cardiomyopathies and channelopathies and more genes are yet to
be discovered, a negative genetic finding does not necessarily exclude a
genetic basis of disease in patients.
Major limitations of the current study include its
small sample size, incomplete family data for co-segregation study, and
lack of functional study of novel variants. We observed a lower rate of
use of genetic tests in early years that might have been due to
insufficient awareness among clinicians about the clinical usefulness of
such tests for channelopathies and cardiomyopathies. Clinical indications
published in an expert consensus statement on the state of genetic testing
for channelopathies and cardiomyopathies from the Heart Rhythm Society and
European Heart Rhythm Association provide a good reference to determine
when a genetic test should be requested.5
In our hospital, referral information can be accessed on
http://kwcpath.home/genetics/ and more information about genetic service
provision in public hospitals is available in the Hong Kong Hospital
Authority Genetic Test Formulary (http://gtf.home/). A comprehensive
system of cardiac genetics service is required for an efficient referral
system, resource funding, training, and appropriate long-term follow-up.
Conclusions
We present the phenotypic and genotypic characteristics of 28 unrelated Hong Kong Chinese patients diagnosed across a 10-year period. For each disease entity, it was beyond our reach in the past decade to exhaustively screen for all known genes. We therefore focus on the most common ones when investigating cardiac genetics. Even so, genetic analysis can provide an accurate diagnosis
and is of utmost importance for the management of patients and their families. Non-penetrance, variable expressivity, phenotype-genotype correlation, susceptibility risk, and digenic inheritance have been reported. Genetic testing also allows for genetic counselling on the recurrence risk. Correct interpretation of genetic findings for careful genetic counselling requires professional expertise with relevant experience in both clinical medicine and molecular genetics. Next-generation sequencing will improve diagnostic performance in this genetically heterogeneous group of channelopathies and cardiomyopathies, and could become a mainstay diagnostic tool.
Author contributions
All authors have made substantial contributions to the concept or design of this study; acquisition of data; analysis or interpretation of data; drafting of the article; and critical revision for important intellectual content.
Funding/support
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Declaration
All authors have no conflicts of interest to disclose. All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Ethical approval
Local ethical approval of this study was obtained (KW/EX/09-155).
References
1. Herman A, Bennett MT, Chakrabarti S,
Krahn AD. Life threatening causes of syncope: channelopathies and
cardiomyopathies. Auton Neurosci 2014;184:53-9. Crossref
2. Ackerman MJ, Marcou CA, Tester DJ.
Personalized medicine: genetic diagnosis for inherited
cardiomyopathies/channelopathies. Rev Esp Cardiol (Engl Ed)
2013;66:298-307. Crossref
3. Priori SG, Wilde AA, Horie M, et al.
HRS/EHRA/APHRS expert consensus statement on the diagnosis and management
of patients with inherited primary arrhythmia syndromes: document endorsed
by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in
June 2013. Heart Rhythm 2013;10:1932-63. Crossref
4. Authors/Task Force members, Elliott PM,
Anastasakis A, et al. 2014 ESC guidelines on diagnosis and management of
hypertrophic cardiomyopathy: the task force for the diagnosis and
management of hypertrophic cardiomyopathy of the European Society of
Cardiology (ESC). Eur Heart J 2014;35:2733-79. Crossref
5. Ackerman MJ, Priori SG, Willems S, et
al. HRS/EHRA expert consensus statement on the state of genetic testing
for the channelopathies and cardiomyopathies: this document was developed
as a partnership between the Heart Rhythm Society (HRS) and the European
Heart Rhythm Association (EHRA). Europace 2011;13:1077-109. Crossref
6. Medeiros-Domingo A, Bhuiyan ZA, Tester
DJ, et al. The RYR2-encoded ryanodine receptor/calcium release
channel in patients diagnosed previously with either catecholaminergic
polymorphic ventricular tachycardia or genotype negative, exercise-induced
long QT syndrome: a comprehensive open reading frame mutational analysis.
J Am Coll Cardiol 2009;54:2065-74. Crossref
7. Wallis Y, Payne S, McAnulty C, et al.
Practice guidelines for the evaluation of pathogenicity and the reporting
of sequence variants in clinical molecular genetics. Association for
Clinical Genetic Science; 2013.
8. Adam MP, Ardinger HH, Pagon RA, Wallace
SE, editors. GeneReviews [internet]. Seattle (WA): GeneReviews; 1993.
Available from: https://www.ncbi.nlm.nih.gov/books/NBK1116/. Accessed 19
Feb 2018.
9. Bokil NJ, Baisden JM, Radford DJ,
Summers KM. Molecular genetics of long QT syndrome. Mol Genet Metab
2010;101:1-8. Crossref
10. Giudicessi JR, Ackerman MJ. Genotype-
and phenotype-guided management of congenital long QT syndrome. Curr Probl
Cardiol 2013;38:417-55. Crossref
11. Priori SG, Napolitano C, Schwartz PJ,
et al. Association of long QT syndrome loci and cardiac events among
patients treated with beta-blockers. JAMA 2004;292:1341-4. Crossref
12. Antzelevitch C, Yan GX, Ackerman MJ,
et al. J-Wave syndromes expert consensus conference report: emerging
concepts and gaps in knowledge. Europace 2017;19:665-94.
13. Antzelevitch C, Yan GX, Ackerman MJ,
et al. J-Wave syndromes expert consensus conference report: emerging
concepts and gaps in knowledge. J Arrhythm 2016;32:315-39. Crossref
14. Furuhashi M, Uno K, Tsuchihashi K, et
al. Prevalence of asymptomatic ST segment elevation in right precordial
leads with right bundle branch block (Brugada-type ST shift) among the
general Japanese population. Heart 2001;86:161-6. Crossref
15. Matsuo K, Akahoshi M, Nakashima E, et
al. The prevalence, incidence and prognostic value of the Brugada-type
electrocardiogram: a population-based study of four decades. J Am Coll
Cardiol 2001;38:765-70. Crossref
16. Sakabe M, Fujiki A, Tani M, Nishida K,
Mizumaki K, Inoue H. Proportion and prognosis of healthy people with coved
or saddle-back type ST segment elevation in the right precordial leads
during 10 years follow-up. Eur Heart J 2003;24:1488-93. Crossref
17. Hiraoka M. Brugada syndrome in Japan.
Circ J 2007;71 Suppl A:A61-8.
18. Juang JM, Tsai CT, Lin LY, et al.
Unique clinical characteristics and SCN5A mutations in patients
with Brugada syndrome in Taiwan. J Formos Med Assoc 2015;114:620-6. Crossref
19. Bezzina CR, Barc J, Mizusawa Y, et al.
Common variants at SCN5A-SCN10A and HEY2 are associated
with Brugada syndrome, a rare disease with high risk of sudden cardiac
death. Nat Genet 2013;45:1044-9. Crossref
20. Behr ER, Savio-Galimberti E, Barc J,
et al. Role of common and rare variants in SCN10A: results from
the Brugada syndrome QRS locus gene discovery collaborative study.
Cardiovasc Res 2015;106:520-9. Crossref
21. Gourraud JB, Barc J, Thollet A, et al.
The Brugada syndrome: a rare arrhythmia disorder with complex inheritance.
Front Cardiovasc Med 2016;3:9. Crossref
22. Juang JJ, Horie M. Genetics of Brugada
syndrome. J Arrhythm 2016;32:418-25. Crossref
23. Priori SG, Napolitano C, Memmi M, et
al. Clinical and molecular characterization of patients with
catecholaminergic polymorphic ventricular tachycardia. Circulation
2002;106:69-74. Crossref
24. Jabbari J, Jabbari R, Nielsen MW, et
al. New exome data question the pathogenicity of genetic variants
previously associated with catecholaminergic polymorphic ventricular
tachycardia. Circ Cardiovasc Genet 2013;6:481-9. Crossref
25. Kawamura M, Ohno S, Naiki N, et al.
Genetic background of catecholaminergic polymorphic ventricular
tachycardia in Japan. Circ J 2013;77:1705-13. Crossref
26. Xiong HY, Alipanahi B, Lee LJ, et al.
RNA splicing. The human splicing code reveals new insights into the
genetic determinants of disease. Science 2015;347:1254806. Crossref
27. Zou Y, Song L, Wang Z, et al.
Prevalence of idiopathic hypertrophic cardiomyopathy in China: a
population-based echocardiographic analysis of 8080 adults. Am J Med
2004;116:14-8. Crossref
28. Frank-Hansen R, Page SP, Syrris P,
McKenna WJ, Christiansen M, Andersen PS. Micro-exons of the cardiac myosin
binding protein C gene: flanking introns contain a disproportionately
large number of hypertrophic cardiomyopathy mutations. Eur J Hum Genet
2008;16:1062-9. Crossref
29. Millat G, Bouvagnet P, Chevalier P, et
al. Prevalence and spectrum of mutations in a cohort of 192 unrelated
patients with hypertrophic cardiomyopathy. Eur J Med Genet 2010;53:261-7.
Crossref
30. Waldmuller S, Muller M, Rackebrandt K,
et al. Array-based resequencing assay for mutations causing hypertrophic
cardiomyopathy. Clin Chem 2008;54:682-7. Crossref
31. Liu X, Jiang T, Piao C, et al.
Screening mutations of MYBPC3 in 114 unrelated patients with
hypertrophic cardiomyopathy by targeted capture and next-generation
sequencing. Sci Rep 2015;5:11411. Crossref
32. Narula N, Favalli V, Tarantino P, et
al. Quantitative expression of the mutated lamin A/C gene in patients with
cardiolaminopathy. J Am Coll Cardiol 2012;60:1916-20. Crossref
33. Vytopil M, Benedetti S, Ricci E, et
al. Mutation analysis of the lamin A/C gene (LMNA) among patients
with different cardiomuscular phenotypes. J Med Genet 2003;40:e132. Crossref
34. Yuan WL, Huang CY, Wang JF, et al.
R25G mutation in exon 1 of LMNA gene is associated with dilated
cardiomyopathy and limb-girdle muscular dystrophy 1B. Chin Med J (Engl)
2009;122:2840-5.
35. Sen-Chowdhry S, Syrris P, Prasad SK,
et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized
clinical entity. J Am Coll Cardiol 2008;52:2175-87. Crossref
36. Semsarian C, Hamilton RM. Key role of
the molecular autopsy in sudden unexpected death. Heart Rhythm
2012;9:145-50. Crossref
37. Torkamani A, Muse ED, Spencer EG, et
al. Molecular autopsy for sudden unexpected death. JAMA 2016;316:1492-4. Crossref