Hong Kong Med J 2018 Jun;24(3):285–92 | Epub 21 May 2018
DOI: 10.12809/hkmj187245
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
REVIEW ARTICLE CME
Jaundice in infants and children: causes, diagnosis,
and management
YY Chee, MB, BS, FHKAM (Paediatrics)1;
Patrick HY Chung, MB, BS, FHKAM (Surgery)2; Rosanna MS Wong,
MB, BS, FHKAM (Paediatrics)1; Kenneth KY Wong, PhD, FHKAM
(Surgery)2
1 Department of Paediatrics and
Adolescent Medicine, The University of Hong Kong, Pokfulam, Hong Kong
2 Department of Surgery, The University
of Hong Kong, Pokfulam, Hong Kong
Corresponding author: Dr Kenneth KY Wong (kkywong@hku.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Jaundice is caused by an accumulation of
bilirubin in the blood. The presentation in infants and children can be
indicative of a wide range of conditions, with some self-limiting and
others potentially life-threatening. This article aims to provide a
concise review of the common medical and surgical causes in children and
discuss their diagnosis and management.
Introduction
Jaundice is caused by the accumulation of bilirubin
in the blood. It can be a result of overproduction of or failure to
metabolise and excrete bilirubin. The incidence of infantile jaundice is
approximately 1 in 2500 to 5000 live births1
2 with a variety of underlying
diagnoses ranging from self-limiting breast milk jaundice to aggressive
life-threatening diseases such as biliary atresia (BA) and liver failure.
Although the clinical features of certain diseases are obvious, some may
have more subtle presentations that necessitate a high index of suspicion
for diagnosis. In general, the differential diagnoses of jaundice in
infancy follow those of adults and can broadly be divided into
pre-hepatic, hepatic, and post-hepatic causes. In some cases, specific
treatment may not be necessary but more often timely management is
required for an optimal outcome. In this article, we highlight several
medical and surgical diagnoses of infantile jaundice and a diagnostic
strategy based on current evidence.
Medical diseases
Breast milk jaundice
Breast milk jaundice was first described more than
50 years ago, with benign unconjugated hyperbilirubinaemia associated with
breastfeeding.3 4 5 It is the
most common cause of prolonged jaundice in an otherwise healthy breastfed
infant born at term. It usually presents in the first 2 to 3 weeks of life
(incidence has been reported as 34%),6
and can persist for as long as 12 weeks before spontaneous resolution.
Total serum bilirubin levels in breast milk jaundice alone do not exceed
200 μmol/L. Diagnosis of breast milk jaundice requires the exclusion of
other possible pathological causes. Table 1 shows the essential clinical features of
breast milk jaundice.

Table 1. Clinical features of breast milk jaundice
The aetiology of breast milk jaundice is not clear.
Animal models suggest that mature breast milk may enhance bilirubin uptake
in the gastrointestinal tract, thus increasing enterohepatic circulation
and unconjugated bilirubin levels.7
8 Higher levels of epidermal growth
factor both in the serum and breast milk of affected infants may offer a
plausible mechanism for breast milk jaundice in the same way.9 Activity of beta-glucuronidase (which deconjugates
intestinal bilirubin) that is higher in human milk than formula milk will
again increase the serum bilirubin level by increasing enterohepatic
circulation.10
Severity and duration of breast milk jaundice may
be affected by a concurrent neonatal manifestation of Gilbert syndrome
which will be discussed later.
Infants with breast milk jaundice require no
treatment provided they are clinically well and the total serum bilirubin
concentration remains below the recommended phototherapy level. The
interruption of breastfeeding is not advised. If total serum bilirubin
exceeds 200 μmol/L, further investigation is required. In case of a
negative workup, the possibility of the additional presence of a mutation
of the hepatic enzyme UGT1A1 (uridine diphosphate glucuronosyltransferase
1A1) conjugating bilirubin in the hepatocyte, ie, Gilbert syndrome, should
be considered. Follow-up (every 2 weeks) should be offered preferably
until a decreasing trend of jaundice becomes evident.
Glucose-6-phosphate dehydrogenase deficiency
Glucose-6-phosphate dehydrogenase (G-6-PD) is an
enzyme found in all cells of the body. Reactive oxygen species (ROS) are
continually formed in the body causing tissue oxidation, disruption of
lipid membranes, destruction of cell enzyme functions, alteration of DNA
structure, and eventually cell death. Glucose-6-phosphate dehydrogenase
plays a major role in neutralising the ROS and offers protection against
tissue oxidative damage. Red blood cells are particularly susceptible to
oxidative stress so the major effect of G-6-PD deficiency is
haematological.
Glucose-6-phosphate dehydrogenase deficiency is a
genetic condition with an X-linked recessive inheritance. Males are more
likely to be affected. In Hong Kong, there is routine cord blood screening
for G-6-PD deficiency and an incidence of around 4.5% in males and 0.5% in
females.11
Glucose-6-phosphate dehydrogenase deficiency–
associated neonatal hyperbilirubinaemia can manifest in two forms: severe
jaundice resulting from acute haemolysis or gradual onset jaundice. Some
G-6-PD–deficient neonates may develop severe haemolysis that results in
rapidly rising serum total bilirubin levels, with the potential to develop
kernicterus, with or without the identification of a known trigger of
haemolysis.12 13 In contrast to the severe haemolytic jaundice,
gradual onset jaundice is less severe and is associated with a slower
increase in total serum bilirubin concentration.
Apart from haemolysis (as evidenced by a falling
haemoglobin with elevated reticulocyte count), diminished bilirubin
clearance plays a role in the pathogenesis of jaundice in G-6-PD
deficiency infants. Serum conjugated bilirubin studies indicate diminished
bilirubin conjugation in G-6-PD–deficient neonates,14 with impaired excretion of conjugated bilirubin into
the small intestine in bile.
Prevention of hyperbilirubinaemia and kernicterus
in G-6-PD–deficient neonates is possible. Parents of neonates affected
should be counselled on the risks of jaundice. They should be advised to
avoid triggers of haemolysis (Table 211).
Predischarge measurement of the bilirubin level using transcutaneous
bilirubin or serum total bilirubin should be performed followed by earlier
and more frequent follow-up.15

Gilbert syndrome
Gilbert syndrome is the most common inherited
disorder of bilirubin glucuronidation. The prevalence of Gilbert syndrome
has been reported to be 5% to 10% in the Caucasian population,16 17 with a
similar prevalence (3-7%) in Chinese.18
19 Uridine diphosphate
glucuronosyltransferase 1A1 is the hepatic enzyme responsible for
bilirubin conjugation. Gilbert syndrome results from a mutation in the UGT1A1
gene promotor region. It manifests only in people who are homozygous for
the genetic mutation, consistent with an autosomal recessive inheritance.
Such mutation may produce structural or functional enzymatic deficiencies,
possibly resulting in impaired bilirubin conjugation and
hyperbilirubinaemia. Such genetic mutations have been demonstrated in
Asian populations.18 19 20
Patients typically present during the adolescent
period (with recurrent episodes of jaundice that may be triggered by
dehydration, fasting, intercurrent illness, menstruation, etc) when
alterations in sex steroid concentrations affect bilirubin metabolism.
Nonetheless they may also present with prolonged breast milk jaundice due
to concurrent Gilbert syndrome. The diagnosis is made by excluding other
causes of unconjugated hyperbilirubinaemia although genetic testing is
available. No treatment is necessary. The long-term outcome is similar to
that for the general population. Nonetheless the Gilbert genotype is
associated with an increased severity and duration of neonatal jaundice.21 22
Viral hepatitis
Among the viral aetiologies in the developing
world, hepatitis A, B, and E are the most common causes of paediatric
acute liver failure (PALF). In the developed world, viruses like herpes
simplex virus (HSV) and enterovirus are more commonly identified as the
aetiological agent.
Hepatitis viruses
Hepatitis A virus infection resulting in PALF is
uncommon in developed countries (2.5% in a PALF registry in North America
and United Kingdom).23 Nonetheless
acute hepatitis A virus infection accounts for up to 80% of PALF cases in
developing countries.24 Similarly,
acute hepatitis B virus (HBV) infection causing PALF is uncommon in the
West where HBV is not endemic. On the contrary, in areas where HBV is
endemic, it accounts for up to 46% of PALF.25
Hepatitis E virus infection has rarely been identified as the cause of
PALF. Pregnant women have a high risk of fulminant hepatitis associated
with hepatitis E virus infection, with a particularly high risk during the
third trimester of pregnancy. The risk of symptomatic hepatitis in the
newborn is high if a pregnant woman acquires hepatitis E virus infection
during pregnancy.
Infection with viruses other than hepatitis viruses
Herpes simplex virus should be considered an
important and treatable cause of PALF. Herpes simplex virus most commonly
affects infants and newborns. In a registry study from North America and
the UK, HSV was identified in 25% of young infants (0-6 months) with PALF.23
Other viruses associated with PALF include
enterovirus and Epstein-Barr virus. The Enterovirus family (including
echovirus, Coxsackie A and B virus) was identified as the aetiological
agent of acute liver failure in 2.7% of young infants (0-90 days of age)
in a multi-centre registry in North America and the UK.26 Epstein-Barr virus is more frequently implicated in
PALF in older children and adolescents.
Chinese herbal medicine–associated hepatotoxicity
Chinese herbs are being increasingly used in the
paediatric population in certain parts of the world and some have been
implicated in the development of hepatotoxicity. A recent retrospective
review of Chinese herbal medicine–induced liver injury in Beijing, China
revealed that Ephedra sinica and Polygonum multiflorum
were the major culprit herbs.27
These ingredients are found in products such as Gan-mao soft capsules
(感冒軟膠), Xiao-er-ke-chuan-ling (小兒咳喘靈) granules, and Shou-wu-yan-shou
(首烏延壽丹) tablets that are used to treat upper respiratory tract infection
or vitiligo. Jaundice is the most common clinical sign. Median days from
herbal ingestion to appearance of presenting symptoms can be up to 30
days. Therefore, it is important to enquire about the use of traditional
Chinese medicine in the weeks preceding clinical presentation.
Surgical diseases
Surgical jaundice commonly refers to obstructive
jaundice and consequent impaired biliary drainage. Unlike adults, the
causes of obstruction in infants are usually congenital. The
differentiation from medical causes can be made by measuring the level of
conjugated bilirubin (which will usually be raised in cases of obstructive
jaundice) as well as examination of the biliary system by ultrasound scan.
Obstructive jaundice is curable by surgery but the magnitude of surgery
ranges from minor to ultra-major. The management of individual diagnoses
will be discussed below.
Biliary atresia
The first description in the English language of a
condition similar to BA appeared in a textbook written by Dr John Burns
from the University of Glasgow in 1817.28
Nonetheless it was more than a century later before the first operation
was performed by Dr William Ladd from Boston in an attempt to correct BA.29 Unfortunately, his surgery did
not improve the outcome of this condition and BA was at this time regarded
as ‘the darkest chapter in paediatric surgery’. In 1959, Dr Morio Kasai
from Japan reported his radical surgery for BA with a higher success rate.30 It was only then that BA became
a potentially curable condition and the operation that was named after Dr
Kasai is now the standard surgical approach for BA.
Biliary atresia is a rare disorder with an
incidence that varies widely among populations (1 in 5000 in Asians to 1
in 18 000 in Caucasians). In 10% to 20% of patients, the disease is
associated with other congenital anomalies.31
The disease is characterised by inflammatory sclerosing cholangiopathy
affecting the entire biliary tract. The bile duct is replaced by fibrous
tissue without luminal patency (Fig 1). Its aetiology remains largely unknown and
the most widely accepted theory is that unknown exogenous factors trigger
a series of self-limiting inflammatory events in a genetically predisposed
individual during the embryonic or perinatal period. Current evidence
suggests that genetics play an important role in the pathogenesis of BA.32 A genome-wide association study
conducted by a group of scientists in Hong Kong discovered a BA-associated
region on chromosome 10q24.2 that could alter the expression of adducin 3
in the liver.33 Nonetheless
genetic abnormality alone cannot be the sole explanation since BA is not
an inherited disease.

Figure 1. An intra-operative photograph showing a 2-month-old boy with biliary atresia, with the entire biliary tract replaced by fibrotic tissue (arrow) without luminal patency
Antenatal diagnosis is difficult and only a few
small case series have been published.34
Affected babies usually present with prolonged jaundice beyond the
neonatal period. Due to the absence of bile pigment in the stool, the
stool is typically pale in colour (Fig 2). The passage of pale-coloured stool from a
young infant should always raise the suspicion of BA. Liver function tests
will reveal a cholestatic pattern that may help to differentiate from
other causes of infantile jaundice. The gallbladder will be absent or
small on ultrasound scan. At a later stage, the liver may demonstrate
parenchymal or fibrotic changes. Although radio-isotope scan, such as a
techetium-99m ethyl hepatic iminodiacetic acid scan may show impaired
biliary excretion, this finding is not confirmatory. The diagnosis of BA
should be confirmed by direct visualisation of the fibrotic biliary tract.
If necessary, an intra-operative cholangiogram can be performed and is
considered normal only if there is passage of contrast up to the
intrahepatic ducts as well as down to the duodenum. This diagnostic
procedure can now be performed via a laparoscopic approach.
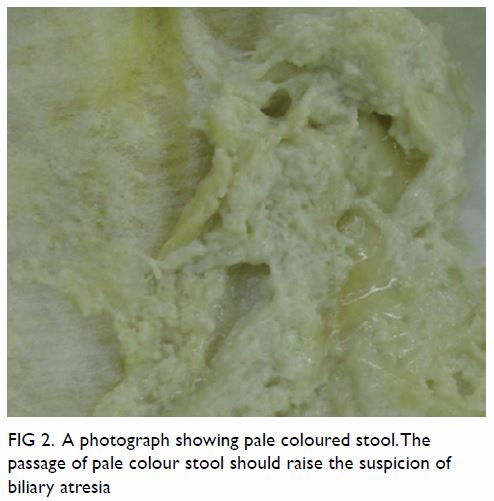
Figure 2. A photograph showing the passage of pale-coloured stool should raise the suspicion of biliary atresia
In the majority of cases, the Kasai operation is
still regarded as the operation of choice to treat BA and should be
carried out in a highly specialised centre. Indeed, the outcome of Kasai
operation in the UK has significantly improved following centralisation of
all BA cases to three major centres after 1999.35
The operation consists of the excision of the fibrous cord at the porta
and restoration of biliary drainage by portoenterostomy. Despite an
uneventful operation, jaundice clearance can be achieved in only 60% to
70% of patients and the 5-year native liver survival rate is roughly 50%
only.36 37
Factors that may improve the outcome including the timing of surgery,
experience of the surgeon, surgical approach, and use of adjuvant
medications have been studied extensively but a definitive answer is still
lacking. Recurrent cholangitis has been found to be associated with a poor
outcome and therefore each cholangitic episode should be treated
aggressively.38 About 30% to 40%
of post-Kasai patients will eventually develop end-stage liver failure and
require liver transplantation as the salvage treatment.39
Alagille syndrome
Alagille syndrome is an autosomal dominant disease
with a variable penetrance. The disorder is believed to be caused by a
defect in the Notch signalling pathway that is important for normal
embryonic development.40 This
syndrome may present with infantile jaundice resembling BA but it also
commonly affects other systems including the cardiovascular,
musculoskeletal, and ocular systems. Some patients will have a
characteristic facial expression (broad forehead, deep-set eyes and
pointed chin). The manifestation in the hepatobiliary system is
characterised by the paucity of intrahepatic bile ducts resulting in
cholestatic jaundice during infancy. The diagnosis is sometimes confused
with BA. It is not uncommon for the Kasai operation to be performed on a
patient with Alagille syndrome with an invariably poor outcome.
Preoperative distinction from BA is possible by genetic testing for JAG1
mutations but this mutation is also found in some patients with BA,
leading to the belief that Alagille syndrome and BA belong to the same
spectrum of disease.41 At present,
the only effective treatment to correct Alagille syndrome is liver
transplantation.42
Inspissated bile syndrome
Inspissated bile syndrome describes a condition
where the bile duct is obstructed due to the impaction of a thick bile
plug or sludge during the neonatal or infantile period. It is sometimes
associated with prematurity, cystic fibrosis, or prolonged use of total
parenteral nutrition but it can occur without an obvious underlying cause.
The use of fluconazole has been reported as a risk factor for developing
this disorder.43 The affected
patient will present with symptoms of obstructive jaundice. Ultrasound
examination may occasionally reveal the presence of sludge in the biliary
system and dilatation of the bile duct. When the obstruction is mild, the
bile plug may dissolve with hydration and high-dose ursodeoxycholic acid.
Nonetheless in severe cases with prolonged obstruction, biliary
obstruction may lead to liver damage and cirrhosis. Inspection of the
gallbladder and the entire biliary tract should be performed to exclude
other causes of obstructive jaundice. At the same time, an operative
cholangiogram can be carried out to exclude BA by showing the passage of
contrast into the intrahepatic ducts as well as small bowel (Fig
3). It also serves a therapeutic purpose by dissolving the bile
plug. Some surgeons advocate concomitant cholecystectomy but it is not
always necessary when the diagnosis is straightforward.
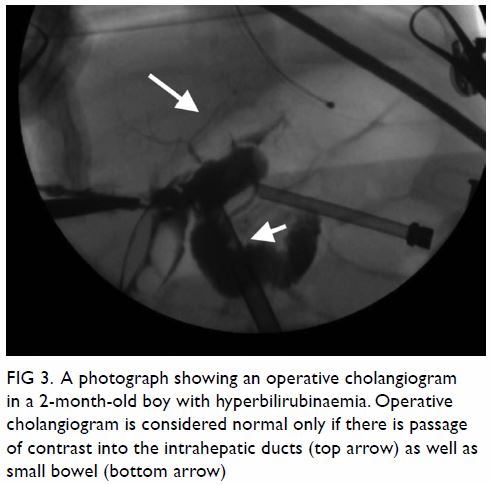
Figure 3. A photograph showing an operative cholangiogram in a 2-month-old boy with hyperbilirubinaemia. Operative cholangiogram is considered normal only if there is passage of contrast into the intrahepatic ducts (top arrow) as well as small bowel (bottom arrow)
Choledochal cyst
Choledochal cyst is a congenital disorder
characterised by cystic dilatation of the intrahepatic and/or extrahepatic
bile duct. The estimated incidence is around 1 in 5000 live births and
slightly more in Asians.44 The
diagnosis is usually made in the first few years of life when the patient
presents with jaundice or abdominal pain. In recent years, antenatal
diagnosis has become more common and more cysts are detected on prenatal
scans. Occasionally, the disease can remain asymptomatic until adulthood
when it presents with cholangitis. Malignant transformation into
cholangiocarcinoma is a rare but possible sequelae of untreated
choledochal cyst and thus, surgical excision is recommended.45
Choledochal cyst is traditionally classified into
five types according to the Todani classification with type I cyst being
the most common.46 Apart from a
stenotic opening at the distal common bile duct causing biliary
obstruction, the abnormal union of the pancreatic duct with a long common
channel can predispose the reflux of pancreatic juice into the bile duct.
Antenatally diagnosed choledochal cyst does not require fetal intervention
and asymptomatic cysts after birth can be observed for a while.
Nonetheless the observation period should not be long to avoid
cholangitis. Earlier surgery has been found to be associated with less
liver injury as well as operative complications.47
Cholangitic episodes should be treated with potent antibiotics and biliary
drainage by either percutaneous or operative means if necessary to avoid
progression to life-threatening sepsis.
Complete cyst excision and hepaticojejunostomy is
regarded as the standard operative treatment for choledochal cyst. Since
the first report of successful laparoscopic surgery in 1995,48 most centres now perform laparoscopic cyst excision (Fig 4). Previous studies have demonstrated superior
outcomes compared with open surgery and it is safe and feasible in young
infants. With the advances in laparoscopic techniques among paediatric
surgeons, an even more minimally invasive approach by single-incision
laparoscopic surgery has been adopted in some centres with satisfactory
results.49
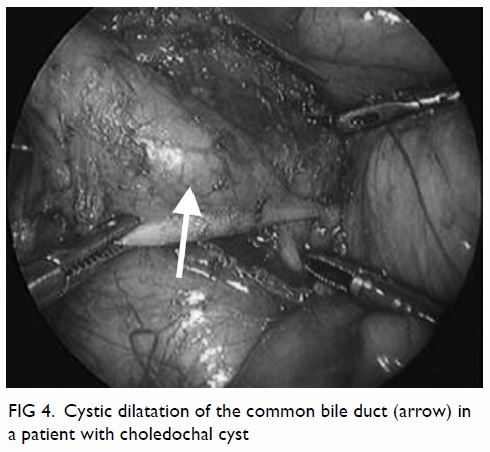
Figure 4. Cystic dilatation of the common bile duct (arrow) in a patient with choledochal cyst
Diagnostic algorithm
A diagnostic algorithm for infantile jaundice is
summarised in Figure 5. Patients with jaundice beyond the neonatal
period require a thorough evaluation for underlying causes. This should
always start with recording a careful history of the antenatal and
perinatal periods including prenatal ultrasonography findings, G-6-PD
status, result of the newborn metabolic screen, etc. It is necessary to
obtain both the child and mother’s medication history to identify any
potential hepatotoxic agents. Although breast milk jaundice is a common
cause of infantile jaundice, other aetiologies should be considered
especially if the infant is not gaining weight, if the total bilirubin
level exceeds 200 μmol/L, and in the presence of red flag symptoms. The
passage of pale stool or tea-coloured urine must not be missed. Physical
examination should not be limited to the abdomen and a systematic
examination should be carried out to look for associated anomalies. Stool
can be saved for inspection of the colour. Blood tests should include a
complete blood picture (along with reticulocyte count and peripheral blood
smear) to exclude haemolytic diseases. The bilirubin level is measured and
it is essential to distinguish between unconjugated and conjugated
hyperbilirubinaemia as they suggest different disease entities. An
increased level of parenchymal enzyme aspartate aminotransferase/alanine
aminotransferase may suggest liver injury due to virus-/drug-induced
causes or autoimmune diseases. On the contrary, obstructive causes are
suggested by an increased level of ductal enzyme alkaline
phosphatase/gamma glutamyl transpeptidase. Serology and antigen of
hepatitis viruses can be checked by sending blood samples to a
microbiology laboratory. An ultrasound scan can detect the presence of
anatomical anomalies in the biliary tract such as BA or choledochal cyst.
A radioisotope scan will help to confirm the presence of biliary
obstruction but does not always give clues to the underlying diagnosis.
Laparoscopic examination of the biliary tract should be arranged when BA
cannot be excluded from the above investigations. Nonetheless laparoscopy
should also be arranged if cholestasis remains unresolved despite normal
imaging to exclude the possibility of inspissated bile plug syndrome.
Intra-operatively, a cholangiogram can be performed by injecting contrast
into the gallbladder to confirm the patency of the biliary tract. It also
serves the purpose of dissolving any bile plug that may be the cause of
obstruction. A liver biopsy can be performed at the conclusion of the
procedure to determine the degree of liver injury. The paucity of bile
duct in a liver biopsy specimen is suggestive of Alagille syndrome.
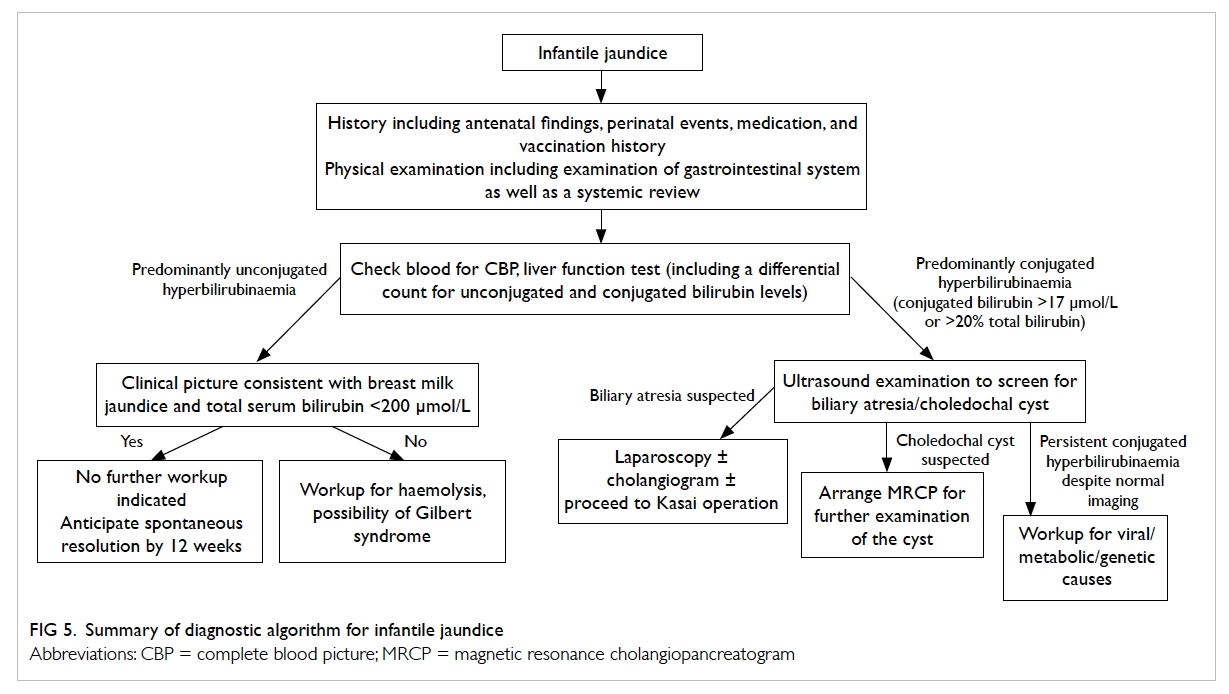
Figure 5. Summary of diagnostic algorithm for infantile jaundice
Conclusion
Infantile jaundice is a common but potentially
life-threatening condition. Referral to a specialist is necessary if
jaundice persists beyond the neonatal period. The differentiation between
medical and surgical causes should be made early on by measuring the blood
level of conjugated and unconjugated bilirubin. Laparoscopy should be
considered in any patient with persistent cholestatic jaundice to exclude
BA that requires early intervention.
Declaration
As an editor of this journal, KKY Wong was not
involved in the peer review process of this article. All other authors
have no conflicts of interest to disclose. All authors had full access to
the data, contributed to the study, approved the final version for
publication, and take responsibility for its accuracy and integrity.
References
1. Suchy FJ. Neonatal cholestasis. Pediatr
Rev 2004;25:388-96.
2. Wang JS, Tan N, Dhawan A. Significance
of low or normal serum gamma glutamyl transferase level in infants with
idiopathic neonatal hepatitis. Eur J Pediatr 2006;165:795-801. Crossref
3. Gartner LM, Arias IM. Studies of
prolonged neonatal jaundice in the breast-fed infant. J Pediatr
1966;68:54-66. Crossref
4. Newman AJ, Gross S. Hyperbilirubinemia
in breast-fed infants. Pediatrics 1963;32:995-1001.
5. Stiehm ER, Ryan J. Breast-milk jaundice.
Report of eight cases and effect of breast feeding on incidence and
severity of unexplained hyperbilirubinemia. Am J Dis Child 1965;109:212-6.
Crossref
6. Maisels MJ, Clune S, Coleman K, et al.
The natural history of jaundice in predominantly breastfed infants.
Pediatrics 2014;134:e340-5. Crossref
7. Alonso EM, Whitington PF, Whitington SH,
Rivard WA, Given G. Enterohepatic circulation of nonconjugated bilirubin
in rats fed with human milk. J Pediatr 1991;118:425-30. Crossref
8. Gartner LM, Lee KS, Moscioni AD. Effect
of milk feeding on intestinal bilirubin absorption in the rat. J Pediatr
1983;103:464-71. Crossref
9. Kumral A, Ozkan H, Duman N, Yesilirmak
DC, Islekel H, Ozalp Y. Breast milk jaundice correlates with high levels
of epidermal growth factor. Pediatr Res 2009;66:218-21. Crossref
10. Gourley GR, Arend RA.
beta-Glucuronidase and hyperbilirubinaemia in breast-fed and formula-fed
babies. Lancet 1986;1:644-6. Crossref
11. Glucose-6-Phosphate Dehydrogenase
(G6PD) Deficiency. DH2289, Clinical Genetic Service, Department of Health,
Hong Kong. 2016. Available from:
http://www.dh.gov.hk/english/main/main_cgs/files/DH2289E%20G6PD%20English.pdf.
Accessed 1 Nov 2017.
12. Valaes T. Severe neonatal jaundice
associated with glucose-6-phosphate dehydrogenase deficiency: pathogenesis
and global epidemiology. Acta Paediatr Suppl 1994;394:58-76. Crossref
13. Kaplan M, Hammerman C. Severe neonatal
hyperbilirubinemia. A potential complication of glucose-6-phosphate
dehydrogenase deficiency. Clin Perinatol 1998;25:575-90, viii. Crossref
14. Kaplan M, Rubaltelli FF, Hammerman C,
et al. Conjugated bilirubin in neonates with glucose-6-phosphate
dehydrogenase deficiency. J Pediatr 1996;128:695-7. Crossref
15. American Academy of Pediatrics
Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in
the newborn infant 35 or more weeks of gestation. Pediatrics
2004;114:297-316. Crossref
16. Owens D, Evans J. Population studies
on Gilbert’s syndrome. J Med Genet 1975;12:152-6. Crossref
17. Sieg A, Arab L, Schlierf G, Stiehl A,
Kommerell B. Prevalence of Gilbert’s syndrome in Germany. Dtsch Med
Wochenschr 1987;112:1206-8. Crossref
18. Long J, Zhang S, Fang X, Luo Y, Liu J.
Neonatal hyperbilirubinemia and Gly71Arg mutation of UGT1A1 gene: a
Chinese case-control study followed by systematic review of existing
evidence. Acta Paediatr 2011;100:966-71. Crossref
19. Chang PF, Lin YC, Liu K, Yeh SJ, Ni
YH. Prolonged unconjugated hyperbilirubinemia in breast-fed male infants
with a mutation of uridine diphosphate-glucuronosyl transferase. J Pediatr
2009;155:860-3. Crossref
20. Maruo Y, Morioka Y, Fujito H, et al.
Bilirubin uridine diphosphate-glucuronosyltransferase variation is a
genetic basis of breast milk jaundice. J Pediatr 2014;165:36-41.e1. Crossref
21. Roy-Chowdhury N, Deocharan B, Bejjanki
HR, et al. Presence of the genetic marker for Gilbert syndrome is
associated with increased level and duration of neonatal jaundice. Acta
Paediatr 2002;91:100-1. Crossref
22. Monaghan G, McLellan A, McGeehan A, et
al. Gilbert’s syndrome is a contributory factor in prolonged unconjugated
hyperbilirubinemia of the newborn. J Pediatr 1999;134:441-6. Crossref
23. Schwarz KB, Dell Olio D, Lobritto SJ,
et al. Analysis of viral testing in nonacetaminophen pediatric acute liver
failure. J Pediatr Gastroenterol Nutr 2014;59:616-23. Crossref
24. Moreira-Silva SF, Frauches DO, Almeida
AL, Mendonça HF, Pereira FE. Acute liver failure in children: observations
in Vitória, Espírito Santo State, Brazil. Rev Soc Bras Med Trop
2002;35:483-6. Crossref
25. Chen HL, Chang CJ, Kong MS, et al.
Pediatric fulminant hepatic failure in endemic areas of hepatitis B
infection: 15 years after universal hepatitis B vaccination. Hepatology
2004;39:58-63. Crossref
26. Sundaram SS, Alonso EM, Narkewicz MR,
et al. Characterization and outcomes of young infants with acute liver
failure. J Pediatr 2011;159:813-8.e1. Crossref
27. Zhu Y, Li YG, Wang JB, et al. Causes,
features, and outcomes of drug-induced liver injury in 69 Children from
China. Gut Liver 2015;9:525-33. Crossref
28. Burns J. The Principals of Midwifery:
Including the Disease of Women and Children. London: Longman; 1817.
29. Ladd WE. Congenital atresia and
stenosis of the bile ducts. JAMA 1928;91:1082-5. Crossref
30. Kasai M, Suzuki S. A new operation for
“non-correctable” biliary atresia: hepatic portoenterostomy. Shujutsu
1959;(13):733-9.
31. Hartley JL, Davenport M, Kelly DA.
Biliary atresia. Lancet 2009;374:1704-13. Crossref
32. Lakshminarayanan B, Davenport M.
Biliary atresia: a comprehensive review. J Autoimmun 2016;73:1-9. Crossref
33. Cheng G, Tang CS, Wong EH, et al.
Common genetic variants regulating ADD3 gene expression alter biliary
atresia risk. J Hepatol 2013;59:1285-91. Crossref
34. Caponcelli E, Knisely AS, Davenport M.
Cystic biliary atresia: an etiologic and prognostic subgroup. J Pediatr
Surg 2008;43:1619-24. Crossref
35. Davenport M, Ong E, Sharif K, et al.
Biliary atresia in England and Wales: results of centralization and new
benchmark. J Pediatr Surg 2011;46:1689-94. Crossref
36. Bijl EJ, Bharwani KD, Houwen RH, de
Man RA. The long-term outcome of the Kasai operation in patients with
biliary atresia: a systematic review. Neth J Med 2013;71:170-3.
37. Lampela H, Ritvanen A, Kosola S, et
al. National centralization of biliary atresia care to an assigned
multidisciplinary team provides high-quality outcomes. Scand J
Gastroenterol 2012;47:99-107. Crossref
38. Chung PH, Wong KK, Tam PK. Predictors
for failure after Kasai operation. J Pediatr Surg 2015;50:293-6. Crossref
39. Wang P, Xun P, He K, Cai W. Comparison
of liver transplantation outcomes in biliary atresia patients with and
without prior portoenterostomy: a meta-analysis. Dig Liver Dis
2016;48:347-52. Crossref
40. McDaniell R, Warthen DM, Sanchez-Lara
PA, et al. NOTCH2 mutations cause Alagille syndrome, a
heterogeneous disorder of the notch signaling pathway. Am J Hum Genet
2006;79:169-73. Crossref
41. Dědič T, Jirsa M, Keil R, Rygl M,
Šnajdauf J, Kotalová R. Alagille syndrome mimicking biliary atresia in
early infancy. PLoS One 2015;10:e0143939. Crossref
42. Spinner NB, Leonard LD, Krantz ID.
Alagille Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, et al,
editors. GeneReviews®. Seattle; 1993.
43. Brownschidle S, Zenali M, Potenta S,
Sartorelli K, Sullivan J. Neonatal cholestasis due to biliary
sludge—review and report of a case associated with use of Diflucan. Ann
Clin Pathol 2014;2:1018.
44. Singham J, Schaeffer D, Yoshida E,
Scudamore C. Choledochal cysts: analysis of disease pattern and optimal
treatment in adult and paediatric patients. HPB (Oxford) 2007;9:383-7. Crossref
45. Madadi-Sanjani O, Wirth TC, Kuebler
JF, Petersen C, Ure BM. Choledochal cyst and malignancy: A plea for
lifelong follow-up. Eur J Pediatr Surg 2017 Dec 19. Epub ahead of print. Crossref
46. Todani T, Watanabe Y, Toki A, Morotomi
Y. Classification of congenital biliary cystic disease: special reference
to type Ic and IVA cysts with primary ductal stricture. J Hepatobiliary
Pancreat Surg 2003;10:340-4. Crossref
47. Diao M, Li L, Cheng W. Timing of
surgery for prenatally diagnosed asymptomatic choledochal cysts: a
prospective randomized study. J Pediatr Surg 2012;47:506-12. Crossref
48. Farello GA, Cerofolini A, Rebonato M,
Bergamaschi G, Ferrari C, Chiappetta A. Congenital choledochal cyst:
video-guided laparoscopic treatment. Surg Laparosc Endosc 1995;5:354-8.
49. Son TN, Liem NT, Hoan VX.
Transumbilical laparoendoscopic single-site surgery with conventional
instruments for choledochal cyst in children: early results of 86 cases. J
Laparoendosc Adv Surg Tech A 2014;24:907-10. Crossref