DOI: 10.12809/hkmj144500
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
One too many: intellectual disability secondary to undiagnosed phenylketonuria
Joannie Hui, FRCP (Edin), FRACP1; SC Chong, FHKCPaed, FHKAM (Paediatrics)1; LK Law, PhD, FRCPath2; LK Lee, FHKCPaed, MPH (HK)1; Sandy Chang, RD, BSc (Hons) Nutrition/Dietetics3; Phyllis Yau, RD, PgDip Dietetics3; YP Yuen, FHKCPath, FHKAM (Pathology)2
1 Department of Paediatrics, The Chinese University of Hong Kong, Shatin, Hong Kong
2 Department of Chemical Pathology, The Chinese University of Hong Kong, Shatin, Hong Kong
3 Dietetics Department, Prince of Wales Hospital, Shatin, Hong Kong
Corresponding author: Dr Joannie Hui (joanniehui@cuhk.edu.hk)

Case report
Hyperphenylalaninaemia refers to the clinical
condition characterised by increased amounts of
phenylalanine in blood and other tissues. It can result
from either a deficiency of phenylalanine hydroxylase
(PAH) or defects in synthesis or regeneration of
tetrahydrobiopterin (BH4), a cofactor for PAH (Fig 1).
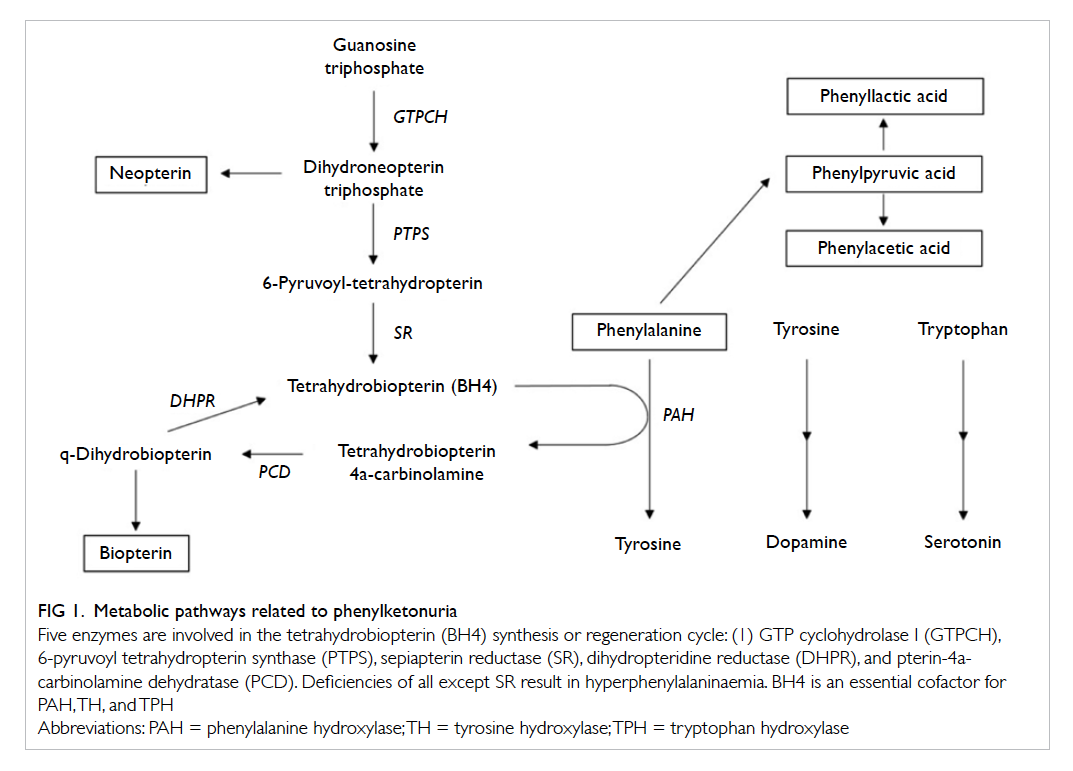
Figure 1. Metabolic pathways related to phenylketonuria
Five enzymes are involved in the tetrahydrobiopterin (BH4) synthesis or regeneration cycle: (1) GTP cyclohydrolase I (GTPCH), 6-pyruvoyl tetrahydropterin synthase (PTPS), sepiapterin reductase (SR), dihydropteridine reductase (DHPR), and pterin-4acarbinolamine dehydratase (PCD). Deficiencies of all except SR result in hyperphenylalaninaemia. BH4 is an essential cofactor for PAH, TH, and TPH
We describe a 2-year-old boy who was
referred by the Maternity Child Health Clinic to the
Department of Paediatrics in June 2014 for assessment
of developmental delay. He was the second child
in his family, born at term and following an uneventful
pregnancy to non-consanguineous Chinese parents.
His birth weight was 3475 g. He was formula-fed
from birth, then gradually weaned to a normal
toddler diet. Family history was unremarkable.
The mother was from Hubei, a province in Central
China, and father was from Shenzhen, a major city
in Southern China. The 5-year-old brother was well
and there were no developmental concerns.
The boy’s early developmental milestones were
unremarkable. He developed social smile at 1 month
of age, sat unsupported at 8 months, and walked
independently at 1 year and 8 months. He started
saying single words at 15 months but at the age of
2 years was still saying only a few single words and
no phrases. Also he was noted to be hyperactive
with behaviour that was at times difficult to
control. His physical growth was satisfactory. His
head circumference was 48.5 cm (50th centile),
weight 14.1 kg (90th centile), and height 87 cm
(50th centile). There were no dysmorphic features
and no abnormalities were detected on physical
examination. His hair was slightly brownish.
Initial baseline investigations revealed normal
blood count and liver, renal, and thyroid function.
Urine organic acid analysis showed markedly
elevated phenylalanine metabolites including
phenyllactic, 3-phenylpyruvic, and phenylacetic
acids. Plasma phenylalanine level was markedly
elevated at 1948 µmol/L (reference range [RR], 26-91 µmol/L) and tyrosine was
47 µmol/L (RR, 24-115 µmol/L).
Urine biopterin was mildly elevated at 5.0 µmol/mmol creatinine (RR, 0.5-3.0) while neopterin
was normal at 1.7 µmol/mmol creatinine (RR, 1.1-4.0). Erythrocyte dihydropteridine reductase activity
was normal. A BH4 loading test was performed to
check for BH4 responsiveness according to standard
protocol.1 Blood for phenylalanine and tyrosine was
checked serially at 8, 16, and 24 hours after each
administration of 20 mg/kg BH4 (Kuvan) orally on
day 1 and day 2 of the loading test. No appreciable
drop in blood phenylalanine level was observed
confirming BH4 non-responsiveness.
All coding exons and flanking introns of the
PAH gene (reference sequence NM_000277.1) were
sequenced using the standard Sanger method. A
heterozygous missense mutation c.860T>C was
detected in exon 8 of the PAH gene. This mutation
changes the highly conserved leucine at position
287 to proline (p.Leu287Pro) [Fig 2]. Another
mutation affecting the same amino acid p.Leu287Gly
has been reported previously in patients with
phenylketonuria (PKU).2 In-silico analyses by four
prediction software (PolyPhen-2, SIFT, Mutation
Taster, PON-P2) also consistently predicted that the
mutation is pathogenic. Therefore, PAH c.860T>C
(p.Leu287Pro) is highly likely to be a pathogenic
mutation. The mother was a carrier of this missense
mutation. PAH gene dosage analysis by multiplex
ligation probe amplification (SALSA MLPA
probemix P055-C1 PAH) did not detect any PAH
gross deletion or duplication. Therefore, the second
PAH mutation of this patient remained unidentified.
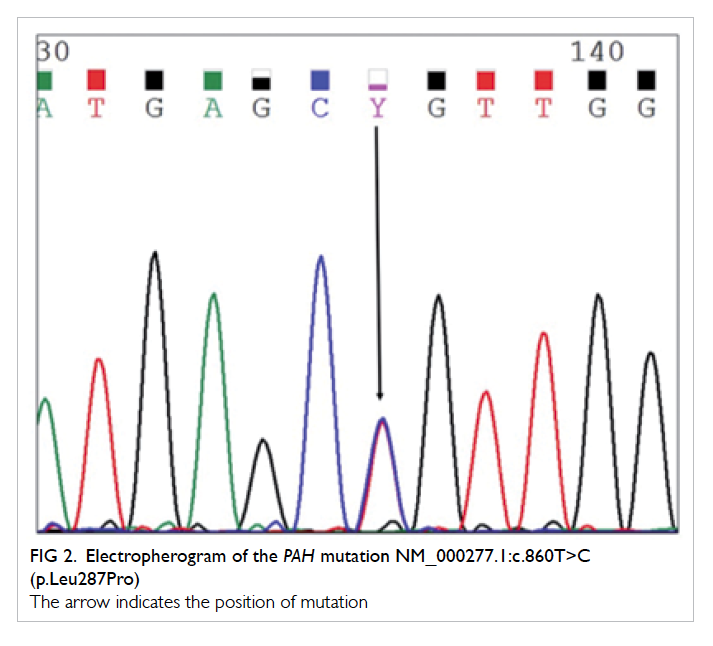
Figure 2. Electropherogram of the PAH mutation NM_000277.1:c.860T>C (p.Leu287Pro)
The arrow indicates the position of mutation
The patient was started on a phenylalaninere-stricted
diet after diagnosis. Special formula
XP-2 powder from SHS was used as the
phenylalanine-free and tyrosine supplement source.
With dietary advice and close supervision, his
phenylalanine levels gradually came down to 300 to 500 µmol/L
while tyrosine levels were maintained at 50 to 80 µmol/L
over the following 6 months. It
is too early to report on his progress in terms of
development and behaviour consequent to better
control of the blood phenylalanine level.
Discussion
We believe this is the first reported case of a
locally born child with classic PKU in Hong Kong.
Although a low phenylalanine diet was commenced
promptly upon diagnosis, the cognitive impairment
that has occurred as a result of the unrecognised
long-standing hyperphenylalaninaemia is likely
irreversible. Other than this patient reported here,
two other classic PKU patients are being followed up
at the authors’ metabolic clinic. Both patients were
born in Guangzhou, the capital city of Guangdong
province in South China. They were diagnosed
through the newborn screening programme in
Guangdong province. With good dietary compliance,
both children have achieved normal growth and
development.
This year marked the 56th anniversary
of PKU newborn screening. Effective newborn
screening programmes worldwide including those
in China have identified thousands of infants
with PKU and prevented intellectual disability
through early diagnosis and treatment. Yet in
Hong Kong the need for PKU screening has never
been seriously addressed. Over the last 30 years,
newborn screening in Hong Kong has remained
unchanged with cord blood screening for G6PD
(glucose-6-phosphate dehydrogenase) deficiency
and congenital hypothyroidism. The current practice
of newborn screening lags behind the rest of the
world, and has never been challenged because for
years there have been only scant cases of 6-pyruvoyl
tetrahydropterin synthase (PTPS) deficiency and no
classic PKU cases reported locally. In the absence of
newborn screening, why has only PTPS deficiency
and no classic PKU patients been identified in Hong
Kong? One reason could be that individuals with
PTPS deficiency often present with more complex
neurological manifestations and are more likely to
undergo extensive investigations. On the contrary,
those with classic PKU present with variable
degrees of intellectual disability and behavioural
problems and no overt neurological signs, and
may not have been as extensively investigated. In
addition, there is a general misconception among
practising clinicians in Hong Kong that PKU is a
disease of the Caucasian population. Even if it does
affect the Chinese population, only the Northern
Chinese are affected. As such, plasma amino acid
or urine organic acid profile may not have been
routinely requested for investigation in children
with unexplained developmental delay, intellectual
disability, behavioural problems, or autistic spectrum
disorders. Further, these investigations may not be
as widely available in non–hospital-based private
laboratories.
Without a territory-wide newborn screening
programme, it is impossible to ascertain the true
incidence of PKU in Hong Kong. Irrespective of
whether Hong Kong does have a different PKU
incidence compared with the rest of China, the
presence of confirmed cases locally provides a strong
argument for diagnosis and treatment of affected
individuals at the earliest instance rather than after
symptomatic presentation. Hong Kong cannot afford
to have more intellectual disability as a result of the
unavailability of PKU screening. Until this programme
becomes universally available, we advocate plasma
amino acid and urine organic acid analysis to be
incorporated into the diagnostic workup for all
children with unexplained developmental delay,
intellectual disability, behavioural problems, and
autistic spectrum disorders.
References
1. Blau N, Hennermann JB, Langenbeck U, Lichter-Konecki U.
Diagnosis, classification, and genetics of phenylketonuria
and tetrahydrobiopterin (BH4) deficiencies. Mol Genet
Metab 2011;104 Suppl:S2-9. Crossref
2. Bardelli T, Donati MA, Gasperini S, et al. Two novel genetic
lesions and a common BH4-responsive mutation of the
PAH gene in Italian patients with hyperphenylalaninemia.
Mol Genet Metab 2002;77:260-6. Crossref