Hong Kong Med J 2015 Dec;21(6):499–510 | Epub 16 Oct 2015
DOI: 10.12809/hkmj144402
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Aetiological bases of 46,XY disorders of sex development in the Hong Kong Chinese population
Angel OK Chan*, MD, FHKAM (Pathology)1;
WM But*, MB, BS, FHKAM (Paediatrics)2;
CY Lee, MB, BS, FHKAM (Paediatrics)3;
YY Lam, MB, BS, FHKAM (Paediatrics)4;
KL Ng, MB, BS, FHKAM (Paediatrics)5;
PY Loung, MB, ChB, FHKAM (Paediatrics)6;
Almen Lam, MB, ChB, FHKAM (Paediatrics)5;
CW Cheng, MSc1;
CC Shek, MB, BS, FRCPath1;
WS Wong, MB, ChB, FHKCPath1;
KF Wong, MD, FHKCPath1;
MY Wong, MB, ChB, FHKAM (Paediatrics)2;
WY Tse, MB, BS, FHKAM (Paediatrics)2
1 Department of Pathology, Queen Elizabeth Hospital, Jordan, Hong Kong
2 Department of Paediatrics, Queen Elizabeth Hospital, Jordan, Hong
Kong
3 Department of Paediatrics and Adolescent Medicine, Caritas Medical
Centre, Shamshuipo, Hong Kong
4 Department of Paediatrics and Adolescent Medicine, Kwong Wah
Hospital, Yaumatei, Hong Kong
5 Department of Paediatrics and Adolescent Medicine, United Christian
Hospital, Kwun Tong, Hong Kong
6 Department of Paediatrics and Adolescent Medicine, Princess Margaret
Hospital, Laichikok, Hong Kong
* AOK Chan and WM But have equal contribution in this study
Corresponding author: Dr Angel OK Chan (cok436@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Objective: Disorders of sex development are due
to congenital defects in chromosomal, gonadal, or
anatomical sex development. The objective of this
study was to determine the aetiology of this group
of disorders in the Hong Kong Chinese population.
Design: Case series.
Setting: Five public hospitals in Hong Kong.
Patients: Patients with 46,XY disorders of
sex development under the care of paediatric
endocrinologists between July 2009 and June 2011.
Main outcome measures: Measurement of serum
gonadotropins, adrenal and testicular hormones,
and urinary steroid profiling. Mutational analysis
of genes involved in sexual differentiation by direct
DNA sequencing and multiplex ligation-dependent
probe amplification.
Results: Overall, 64 patients were recruited for the
study. Their age at presentation ranged from birth
to 17 years. The majority presented with ambiguous
external genitalia including micropenis and severe
hypospadias. A few presented with delayed puberty
and primary amenorrhea. Baseline and post–human
chorionic gonadotropin–stimulated testosterone and
dihydrotestosterone levels were not discriminatory
in patients with or without AR gene mutations. Of
the patients, 22 had a confirmed genetic disease,
with 11 having 5α-reductase 2 deficiency, seven with
androgen insensitivity syndrome, one each with
cholesterol side-chain cleavage enzyme deficiency,
Frasier syndrome, NR5A1-related sex reversal, and
persistent Müllerian duct syndrome.
Conclusions: Our findings suggest that 5α-reductase
2 deficiency and androgen insensitivity syndrome
are possibly the two most common causes of 46,XY
disorders of sex development in the Hong Kong
Chinese population. Since hormonal findings can be
unreliable, mutational analysis of the SRD5A2 and
AR genes should be considered the first-line tests for
these patients.
New knowledge added by this study
- The most common likely causes of 46,XY disorders of sex development (DSD) in our local Chinese population are 5α-reductase 2 deficiency and androgen insensitivity syndrome.
- Blood hormone testing is unreliable in differentiating between androgen insensitivity syndrome and other causes of 46,XY DSD.
- Mutational analysis of the SRD5A2 and AR genes should be considered the first-line investigation in patients with 46,XY DSD.
- When encountering patients with 46,XY DSD, 5α-reductase 2 deficiency and androgen insensitivity syndrome should be considered early as their presence has implications for treatment and prognosis.
Introduction
Disorders of sex development (DSD) are defined
as congenital conditions in which development of
chromosomal, gonadal, or anatomical sex is atypical.1
Traditionally, diagnosis in these patients relies on
extensive endocrine investigation. With advances
in the understanding of the genes involved in sexual
determination and differentiation,2 molecular
diagnosis is playing an increasingly important
role and may even overtake the role of hormonal
assessment as the first-line test, with the latter being
reserved for assessment of disease severity rather
than diagnosis.3
One of the most common causes of 46,XY DSD
in the western population is androgen insensitivity
syndrome (AIS).4 Whether the same is true in our
local population remains unknown. We performed a
prospective multicentre study to explore the possible
aetiological basis of 46,XY DSD in the Hong Kong
Chinese population.
Methods
Patients
Patients who were referred to a paediatric
endocrinologist for the first time or were followed up
in their clinic at five public hospitals in Hong Kong
between July 2009 and June 2011 were recruited
for the study. Inclusion criteria were 46,XY ethnic
Chinese patients who presented with incompletely
virilised, ambiguous, or completely female external
genitalia. Criteria that suggested DSD at birth were
overt genital ambiguity, apparent female genitalia
with an enlarged clitoris, posterior labial fusion,
or an inguinal/labial mass, apparent male genitalia
with bilateral undescended testes, micropenis,
isolated perineal hypospadias, or mild hypospadias
with undescended testes, and discordance between
genital appearance and prenatal karyotype.1
Micropenis is defined as stretched penile length of
<2.5 cm based on the published norm for Chinese.1
Written informed consent was obtained from the
patients and/or parents and the study was approved
by the local ethics committee. None of the patients/parents refused to participate in the study although
seven refused genetic testing (Table 1).
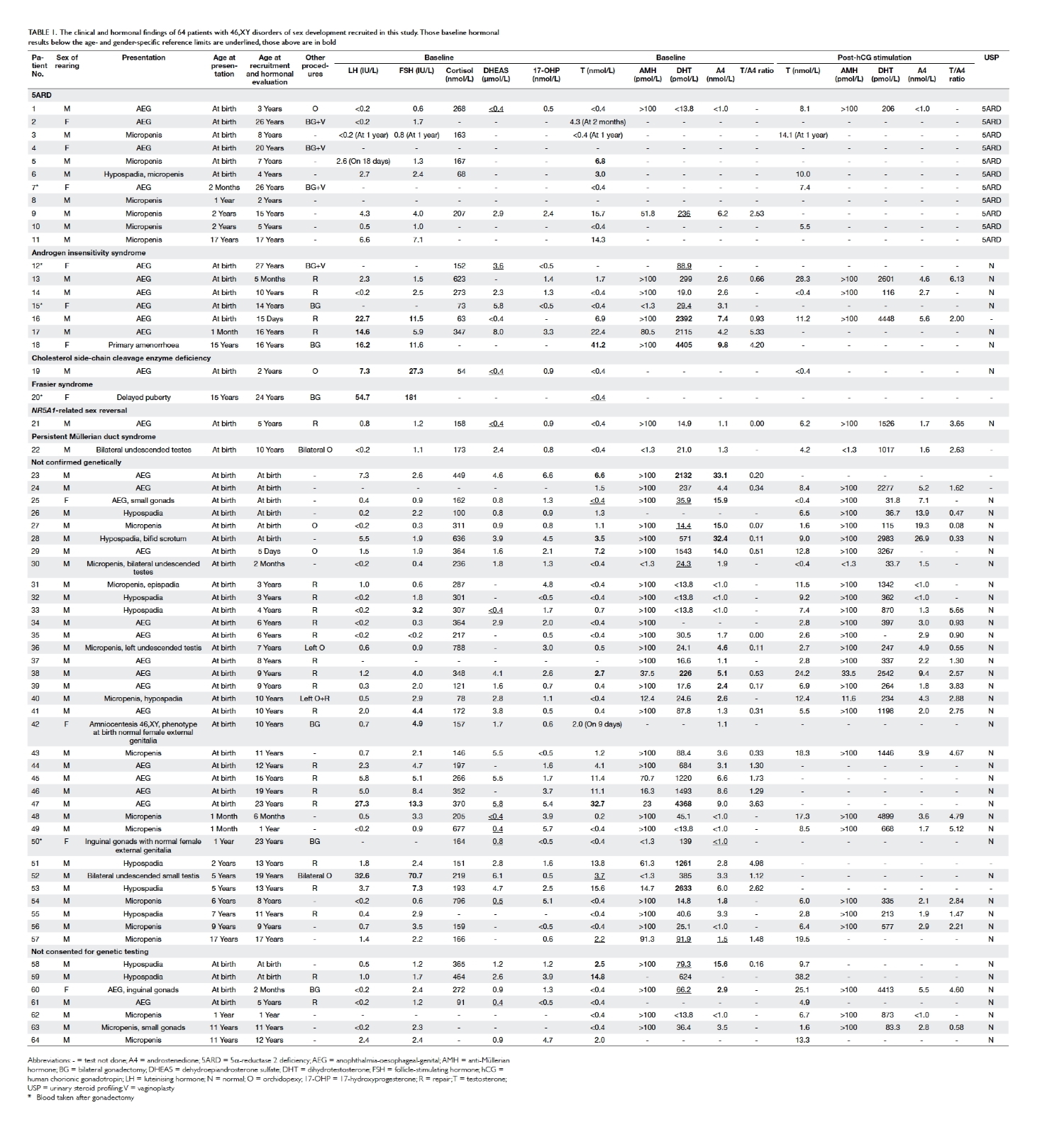
Table 1. The clinical and hormonal findings of 64 patients with 46,XY disorders of sex development recruited in this study. Those baseline hormonal results below the age- and gender-specific reference limits are underlined, those above are in bold
Hormone analysis
Blood was taken from patients for electrolyte
and baseline endocrine assessment and included
measurement of cortisol, 17-hydroxyprogesterone
(17-OHP), dehydroepiandrosterone sulfate,
testosterone (T), androstenedione (A4),
dihydrotestosterone (DHT), anti-Müllerian
hormone (AMH), and gonadotropins. Human
chorionic gonadotropin (hCG) stimulation test was
performed to test for testicular Leydig cell function.
The short synacthen test was also performed when
indicated.
Cortisol, dehydroepiandrosterone sulfate,
and gonadotropins were measured by electro-chemiluminescence
immunoassay (Modular
Analytics E170; Roche, Mannheim, Germany); T
was measured by a competitive immunoenzymatic
assay (ACCESS 2; Beckman Coulter, Brea [CA], US);
17-OHP was measured by liquid chromatography–tandem mass spectrometry using an in-house
method; AMH was measured by an enzyme-linked
immunosorbent assay (AMH Gen II ELISA, A73818;
Beckman Coulter, Brea [CA], US); DHT was measured
by radioimmunoassay (DSL9600i; Beckman Coulter,
Prague, Czech Republic); A4 was measured by
solid-phase competitive chemiluminescent enzyme-labelled
immunoassay (L2KAO2, Immulite 2000;
Siemens, Tarrytown [NY], US). Male reference
intervals were considered the most appropriate for
data interpretation in this study.
Urinary steroid profiling
Spot urine from patients under 3 months of age
and 24-hour urine from those at or older than 3
months of age were processed for steroid profiling as
described previously.5
Molecular analysis
DNA was extracted from peripheral whole blood
using a QIAamp DNA blood kit (Qiagen, Hilden,
Germany). Polymerase chain reaction and direct
DNA sequencing were performed on targeted genes
when suggested by the clinical and hormonal findings.
Otherwise all patients had their AR (androgen
receptor) and NR5A1 (steroidogenic factor 1) genes
sequenced. Those patients with negative genetic
findings were subjected to multiplex ligation-dependent
probe amplification (MLPA) analysis
(P185 Intersex probemix; P074 Androgen Receptor
probemix and P334 Gonadal probemix; MRC-Holland)
to test for gross deletion or gene duplication.
The results were analysed by Coffalyser.Net.
Family genetic studies were performed when
mutation(s) were identified in the index patients.
In-silico analysis for novel missense mutations
The functional effect of novel missense mutations
detected was tested by online in-silico analysis
software SIFT, PolyPhen2, and Align GVGD.
Results
Overall, 64 patients (53 male, 11 phenotypic female),
including 14 new patients, with 46,XY DSD were
recruited into the study. The clinical and hormonal
findings of individual patients are listed in Table 1.
A genetic diagnosis was made in 10 patients prior
to the study. Other major structural abnormalities
were evident in eight (Table 2). Their age at presentation ranged from birth to 17 years. Five
(8%) were born prematurely (24-35 weeks) and
nine (14%) with low birth weight (0.59-2.32 kg). All
had non-consanguineous parents. A family history
of sexual ambiguity was present in six. Overall, 61
(95%) presented with ambiguous external genitalia
including 15 with isolated micropenis, eight
with isolated severe hypospadias, and one with
discordance between the prenatal karyotype and the
postnatal phenotype. Three presented after birth,
one each with inguinal hernia, delayed puberty, and
primary amenorrhoea.

Table 2. Other structural abnormalities detected in eight of the patients in this study
Regarding the hormonal findings, Figure 1 shows the baseline and post-hCG–stimulated T and
DHT levels in patients with mutations detected in
the AR gene and those without, where the results
overlapped between the two groups. Eight patients
(patients 13, 19, 21, 30, 31, 40, 49, and 56) underwent
short synacthen test and with the exception of
patient 19, all had an adequate cortisol response
(>550 nmol/L). Patient 27 had a relatively low
T/A4 ratio before and after hCG stimulation but
sequencing revealed no mutation in his HSD17B3
(17β-hydroxysteroid dehydrogenase III) gene. All
other patients had unremarkable T and A4 levels, as well as T/A4 ratio.
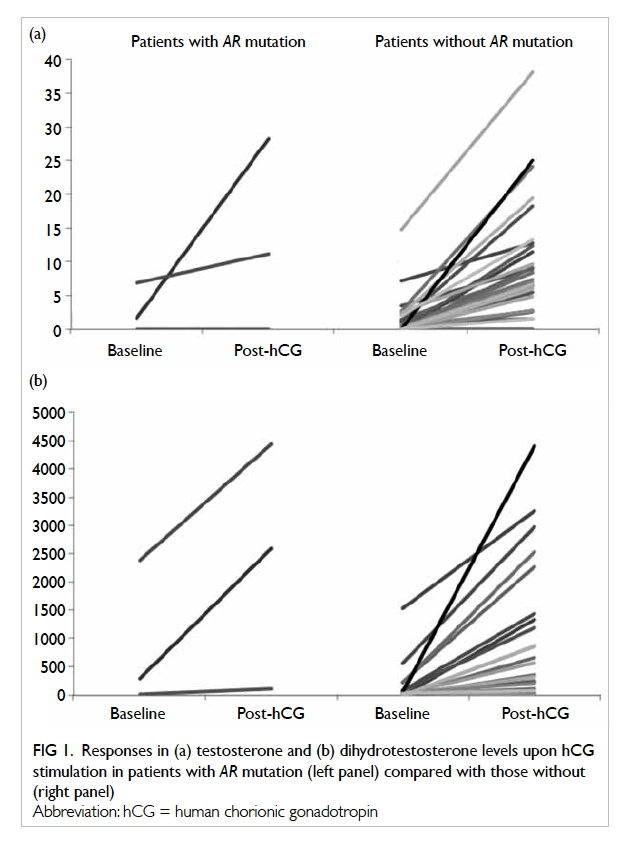
Figure 1. Responses in (a) testosterone and (b) dihydrotestosterone levels upon hCG stimulation in patients with AR mutation (left panel) compared with those without (right panel)
Eleven patients had characteristically low
5α- to 5β-reduced steroid metabolite ratios in
their urine, compatible with the diagnosis of 5α-reductase 2 deficiency (5ARD). This was also
confirmed by mutational analysis of the SRD5A2
(steroid 5α-reductase 2) gene. All other patients had
unremarkable urinary steroid metabolite pattern.
Overall, 22 (39%) patients had a confirmed
genetic diagnosis (Table 3). The most common diagnoses in our cohort were 5ARD (n=11) and AIS
(n=7). Other genetic diagnoses included cholesterol
side-chain cleavage enzyme deficiency (n=1), Frasier
syndrome (n=1), NR5A1-related sex reversal (n=1),
and persistent Müllerian duct syndrome (PMDS;
n=1). The clinical and laboratory findings of patients
19 and 20 have been reported previously.6 7 Patients
12 and 15 had de-novo mutations in the AR gene
and were in mosaic pattern. Patient 21 had a novel
missense variant p.Ala260Val detected in his NR5A1
gene. His AMH level was not low, contrary to some
of the previously reported cases.8 There was also a
clinically significant rise in T level after hCG stimulation.
Short synacthen test demonstrated an adequate
cortisol response (baseline: 720 nmol/L; post–adrenocorticotropin hormone: 822 nmol/L). His
father also carried the same heterozygous mutation
although he denied any symptoms of DSD. This
novel genetic variant was not detected in 100 normal
Chinese subjects (control). Patient 22 had bilateral
undescended testes. He underwent orchidopexy
at the age of 1 year during which the presence of
Müllerian duct structures was suspected. Further
workup including pelvic ultrasound revealed
Müllerian duct structures and extremely low AMH
level. The diagnosis of PMDS was confirmed by
the presence of three heterozygous novel missense
variants in the AMH gene (Tables 3 and 4).
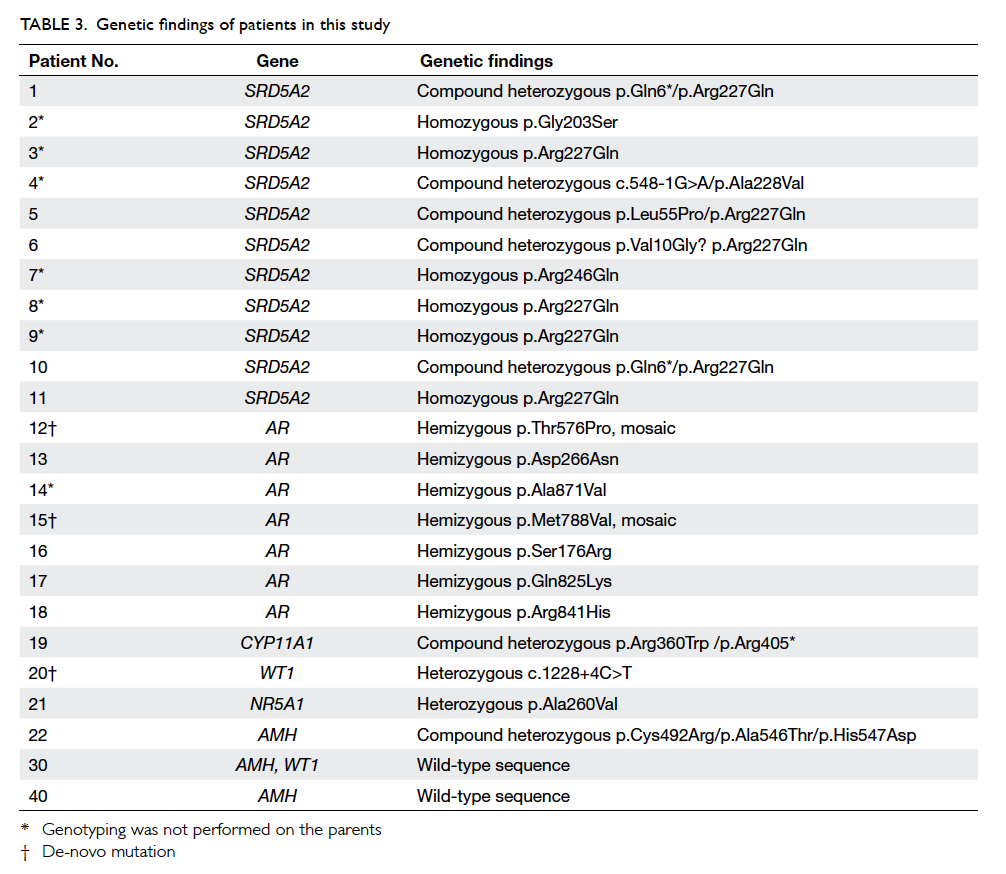
Table 3. Genetic findings of patients in this study
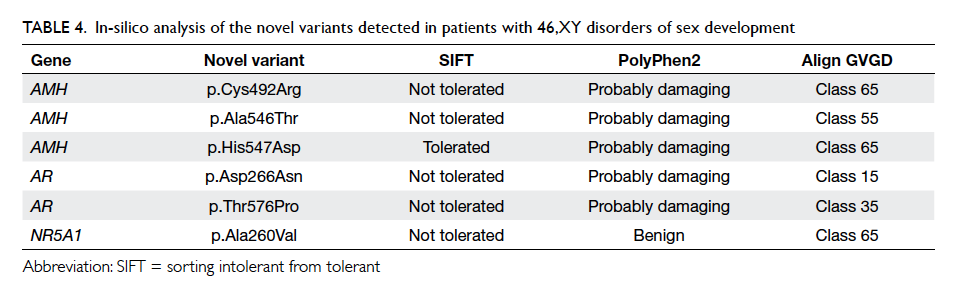
Table 4. In-silico analysis of the novel variants detected in patients with 46,XY disorders of sex development
Six novel genetic variants were identified in the
AMH, AR, and NR5A1 genes (Fig 2). At least two of the three in-silico analysis programmes predicted
the variants to be pathogenic (Table 4). Multiple sequence alignment showed that the amino acids
of concern were highly conserved across different
animal species. All these findings support the
pathogenic nature of these variants accounting for
the patients’ phenotypes.
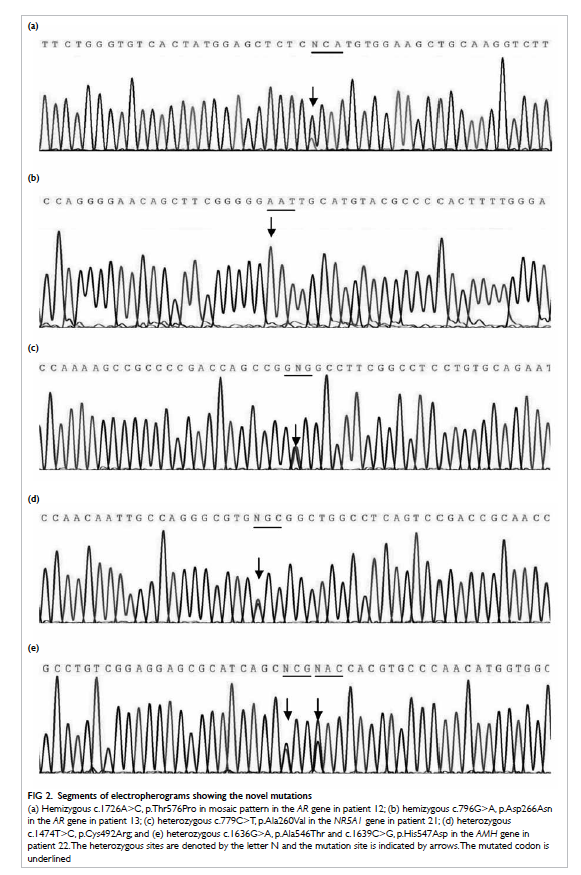
Figure 2. Segments of electropherograms showing the novel mutations
(a) Hemizygous c.1726A>C, p.Thr576Pro in mosaic pattern in the AR gene in patient 12; (b) hemizygous c.796G>A, p.Asp266Asn in the AR gene in patient 13; (c) heterozygous c.779C>T, p.Ala260Val in the NR5A1 gene in patient 21; (d) heterozygous c.1474T>C, p.Cys492Arg; and (e) heterozygous c.1636G>A, p.Ala546Thr and c.1639C>G, p.His547Asp in the AMH gene in patient 22. The heterozygous sites are denoted by the letter N and the mutation site is indicated by arrows. The mutated codon is underlined
Eleven patients were reared as girls because
of severe under-virilisation at birth, including three
with 5ARD, three with AIS, and one with Frasier
syndrome. The underlying genetic causes in the
remaining four patients were undetermined. The
longest follow-up period was 27 years. None of
them has requested change of gender to date. Five
patients (patients 2, 4, 7, 12, and 15) exhibited ‘tom-boy-like’ behaviour during childhood and required
counselling by a clinical psychologist while two
males (patients 17 and 47) requested exogenous T
to augment penile growth after puberty. Patient 20
developed germinoma in her dysgenetic gonad with
no recurrence after surgery.
Discussion
46,XY DSD is a heterogeneous condition caused by
a wide spectrum of disorders. Making an accurate
diagnosis is difficult but important for emergency
medical treatment as some DSDs are associated with
life-threatening Addisonian crisis. In addition, the
diagnosis is essential so that relevant information
and counselling can be provided to parents and
clinical management can be formulated, bearing in
mind the best interests of the child. Initial workup
includes a detailed antenatal and postnatal history,
physical examination, karyotyping, and hormonal
assays. This will guide further workup such as
imaging and genetic analysis. Nonetheless, there are
often limitations to hormonal studies as illustrated
in the present series. The non-distinct pattern of T
and DHT at baseline and following hCG stimulation
in AR mutation–positive and –negative patients
suggest the need to reconsider our laboratory
diagnostic algorithm for AIS.
Androgen insensitivity syndrome is reported
to be the most common cause of 46,XY DSD in a few
ethnic groups,9 10 11 while 5ARD, which is believed to
be rare, was also a major aetiology in our cohort. It
is important to differentiate between 5ARD and AIS
as soon as possible so that patients with 5ARD can
be raised as boys whenever practical.12 The penile
growth of patients with 5ARD can be promoted by
topical DHT treatment and spontaneous virilisation
may occur during puberty. Most of these patients
who are reared as girls during childhood identify
themselves as male and change their gender as
an adult, although we have not received any such
request from our cohort. Exposure to androgen
during the antenatal, postnatal, and pubertal period
may masculinise the brain and influence gender
identity.13 It was found that 5ARD is easy to diagnose
by its characteristic urinary steroid excretion
pattern and its high mutational detection rate in the
SRD5A2 gene.14 Of the 11 patients with 5ARD, eight
harboured the missense mutation p.Arg227Gln in
their SRD5A2 gene, a useful fact to enable screening
for this mutation before proceeding to sequencing
of the whole gene. Unfortunately, patients have
previously been too easily labelled with AIS when
laboratory diagnostic services were less advanced.
This is illustrated by patient 7 who was labelled
as AIS until her urine steroids were analysed and
revealed classic features of 5ARD.15 We recommend
that 5ARD is excluded in all 46,XY DSD patients
before other differential diagnoses are considered.
Moreover, since the baseline and post-hCG–stimulated T and DHT results are unreliable when
diagnosing AIS, genetic study of the AR gene should
also be performed as a first-line investigation.
HSD17B3 deficiency has been reported to be
the most common cause of T biosynthetic defect
leading to 46,XY DSD in some populations, with an
estimated incidence of 1:147 000 in the Netherlands
and as high as 1:200 to 1:300 in Arabians due to their
high consanguinity rate.16 17 Nonetheless, no patient
in our cohort was diagnosed with this condition
based on the hormonal pattern. Ethnic differences
in disease spectrum may be one of the reasons for
this observation. Another possible explanation is
the lack of reliable diagnostic cutoff for the pre- and
post-stimulated T/A4 ratios. George et al18 have
summarised the cutoffs used by various researchers,
with the pre-stimulated cutoff range set at 0.006
to 1.64, and the post-stimulated level set at 0.09 to
3.4 for newborn to teenage groups. The difficulties
in setting up reliable diagnostic cutoffs for the
T/A4 ratio are similar to the T/DHT ratios and have
been discussed in our previous study.14 Furthermore,
HSD17B3 deficiency gives no characteristic findings
on urinary steroid profiling.5 19 Molecular analysis
of the HSD17B3 gene may have offered a means
to diagnose this condition but unfortunately, due
to budget constraints, we were unable to perform
mutational analysis of this gene in all our patients,
although a normal MLPA result in our patients made
gross deletion in this gene unlikely.
The two novel mutations p.Asp266Asn and
p.Thr576Pro in the AR gene lie within the N-terminal
domain of the androgen receptor that is involved
in transcription regulation and DNA binding,
respectively. Missense mutations around these two
codons have been reported in patients with AIS
according to the Androgen Receptor Gene Mutations
Database, April 2013.20 Multiple sequence alignment
shows that both amino acids are highly conserved
among different species, suggesting that aspartic
acid at codon 266 and threonine at codon 576 are
critical for proper receptor function. Similarly, the
alanine at codon 260 of the NR5A1 gene is located in
helix 3 of the ligand-binding domain of the nuclear
receptor,21 and is also a highly conserved region.
Mutation in this region has been reported to result
in 46,XY DSD.8 Replacing alanine at this position
by valine is therefore expected to be deleterious
to the protein function. Phenotypic variability in
NR5A1 gene mutation within a kindred has been
reported and this may explain why patient 21 had
ambiguous external genitalia to such an extent that
he required the attention of a paediatric specialist,
even though his father was fertile, and denied any
symptoms of DSD or need for medical attention.22
For the AMH gene, the 3’ end of exon 5 is one of
the mutational hotspots in patients with PMDS.23
Exon 5 encodes the bioactive C-terminal domain.
The three mutations detected in patient 22 are all
located at highly conserved regions. Although in-vitro
functional characterisation for the mutant
proteins was not performed, the undetectable serum
AMH level in this patient was compatible with
the mutations being pathogenic, possibly due to
abnormal protein folding and increased instability,
as reported previously in mutations located in this
region.24
Gonadal malignancy was rare in our series,
probably because gonadectomy was performed early
in life when the decision of female sex assignment
was made. Although this helps to avoid further
virilisation and to establish gender identity, the
timing of corrective surgery and gonadectomy
remain controversial. Patient advocacy groups have
suggested delaying any surgery for cosmetic reasons
until the patient is mature enough to give informed
consent25 but such practice has not been validated in
our Chinese patients. Whether cultural factors have
any impact on gender assignment remains uncertain
in our community.
Prematurity or low birth weight was not
uncommon in our series. This made diagnosis of
DSD in our patients even more difficult because
ethnic-specific and gestational age– or weight-adjusted anthropometric measurement of the
external genitalia was not available. Assessment of
the genital anatomy relies solely on the experience
of the paediatric specialist and is obviously far from
ideal. A conjoint effort by local paediatricians is
needed to set up these normative data.
Less than half of our patients had a confirmed
diagnosis in the present study. With the increasing
availability of next-generation sequencing technology,
and with its established role in molecular
diagnostic services, including DSD,3 26 it is hoped
that sooner rather than later, most patients will
have a confirmed genetic diagnosis. Nonetheless,
we speculate that some patients have a non-genetic
aetiology since environmental factors may alter the
phenotypic expression. Several animal and human
studies have shown that antenatal exposure to
pesticides and plasticisers may lead to fetal genital
malformation.27
Altogether there was an average of 11 250 male
live births every year in the five public hospitals
that participated in this study. Since 11 newborns
with 46,XY DSD were born in these five hospitals
and were recruited during our study period, this
gives an estimated incidence of 46,XY DSD of
1:2045 male births requiring the input of paediatric
endocrinologists. This figure may underestimate
the true incidence of this group of diseases as some
patients present late and others may have subtle
defects that go unnoticed by our specialists. If the
actual number of patients with chromosomal and
46,XX DSD in our population is considered, the actual
incidence of DSD can be expected to be much higher.
There are a few limitations in this study.
First, the number of patients was relatively small.
This may have resulted in bias in our observation
and the data do not represent the prevalence of
disease in our population. Second, in-vitro study
was not performed on the novel genetic variants for
functional characterisation, although we believe that
all the available evidence indicates the pathogenic
nature of these variants. Third, due to budget
constraints, we were unable to sequence all genes
related to 46,XY DSD.
Conclusions
Our findings indicate that 5ARD and AIS are
possibly the major causes of 46,XY DSD in the Hong
Kong Chinese population. Molecular analyses of
the SRD5A2 and AR genes were demonstrated to
be more reliable than hormonal testing. Since the
missense mutation p.Arg227Gln was a recurrent
hotspot mutation in 5ARD in our local patients, all
patients should be screened for this mutation.
Acknowledgements
We thank Mr YC Ho, Ms YF Wong, and Ms YP Iu for
their technical assistance. The study was supported
by the Queen Elizabeth Hospital Research Grant
2009 QEH/RC/G/0910-A04/R0901 and Kowloon
Central Cluster Research Grant 2012 KCC/RC/G/1213-B01.
References
1. Lee PA, Houk CP, Ahmed SF, Hughes IA; International
Consensus Conference on Intersex organized by the
Lawson Wilkins Pediatric Endocrine Society and the
European Society for Paediatric Endocrinology. Consensus
statement on management of intersex disorders.
International Consensus Conference on Intersex.
Pediatrics 2006;118.e488-500. Crossref
2. Ono M, Harley VR. Disorders of sex development: new
genes, new concepts. Nat Rev Endocrinol 2013;9:79-91. Crossref
3. Arboleda VA, Lee H, Sánchez FJ, et al. Targeted massively
parallel sequencing provides comprehensive genetic
diagnosis for patients with disorders of sex development.
Clin Genet 2013;83:35-43. Crossref
4. Jääskeläinen J. Molecular biology of androgen insensitivity.
Mol Cell Endocrinol 2012;35:4-12. Crossref
5. Chan AO, Shek CC. Urinary steroid profiling in the
diagnosis of congenital adrenal hyperplasia and disorders
of sex development: experience of a urinary steroid referral
centre in Hong Kong. Clin Biochem 2013;46:327-34. Crossref
6. Parajes S, Chan AO, But WM, et al. Delayed diagnosis of
adrenal insufficiency in a patient with severe penoscrotal
hypospadias due to two novel P450 side-chain cleavage
enzyme (CYP11A1) mutations (p.R360W; p.R405X). Eur J
Endocrinol 2012;167:881-5. Crossref
7. Chan WK, To KF, But WM, Lee KW. Frasier syndrome:
a rare cause of delayed puberty. Hong Kong Med J
2006;12:225-7.
8. Allali S, Muller JB, Brauner R, et al. Mutation analysis of
NR5A1 encoding steroidogenic factor 1 in 77 patients
with 46,XY disorders of sex development (DSD) including
hypospadias. PLoS One 2011;6:e24117. Crossref
9. Bangsbøll S, Qvist I, Lebech PE, Lewinsky M. Testicular
feminization syndrome and associated gonadal tumors in
Denmark. Acta Obstet Gynecol Scand 1992;71:63-6. Crossref
10. Boehmer AL, Brinkmann O, Brüggenwirth H, et al.
Genotype versus phenotype in families with androgen
insensitivity syndrome. J Clin Endocrinol Metab
2001;86:4151-60. Crossref
11. Abdullah MA, Saeed U, Abass A, et al. Disorders of sex
development among Sudanese children: 5-year experience
of a pediatric endocrinology clinic. J Pediatr Endocrinol
Metab 2012;25:1065-72. Crossref
12. Mieszczak J, Houk CP, Lee PA. Assignment of the sex of
rearing in the neonate with a disorder of sex development.
Curr Opin Pediatr 2009;21:541-7. Crossref
13. Imperato-McGinley J, Peterson RE, Gautier T, Sturla E.
Androgens and the evolution of male-gender identity
among male pseudohermaphrodites with 5alpha-reductase
deficiency. N Engl J Med 1979;300:1233-7. Crossref
14. Chan AO, But BW, Lee CY, et al. Diagnosis of 5α-reductase
2 deficiency: is measurement of dihydrotestosterone
essential? Clin Chem 2013;59:798-806. Crossref
15. Chan AO, But BW, Lau GT, et al. Diagnosis of 5α-reductase
2 deficiency: a local experience. Hong Kong Med J
2009;15:130-5.
16. Boehmer AL, Brinkmann AO, Sandkuijl LA, et al. 17β-hydroxysteroid
dehydrogenase-3 deficiency: diagnosis,
phenotypic variability, population genetics, and worldwide
distribution of ancient and de novo mutations. J Clin
Endocrinol Metab 1999;84:4713-21. Crossref
17. Rosler A. 17 Beta-hydroxysteroid dehydrogenase 3
deficiency in the Mediterranean population. Pediatr
Endocrinol Rev 2006;3 Suppl 3:455-61.
18. George MM, New MI, Ten S, Sultan C, Bhangoo A. The
clinical and molecular heterogeneity of 17βHSD-3 enzyme
deficiency. Horm Res Paediatr 2010;74:229-40. Crossref
19. Lee YS, Kirk JM, Stanhope RG, et al. Phenotypic variability
in 17β-hydroxysteroid dehydrogenase-3 deficiency and
diagnostic pitfalls. Clin Endocrinol 2007;67:20-8. Crossref
20. Androgen Receptor Gene Mutations Database. Available from: http://androgendb.mcgill.ca/. Accessed Aug 2013.
21. El-Khairi R, Martinez-Aguayo A, Ferraz-de-Souza B, Lin L,
Achermann JC. Role of DAX-1 (NR0B1) and steroidogenic
factor-1 (NR5A1) in human adrenal function. Endocr Dev
2011;20:38-46.
22. Ciaccio M, Costanzo M, Guercio G, et al. Preserved fertility
in a patient with a 46,XY disorder of sex development
due to a new heterozygous mutation in the NR5A1/SF-1
gene: evidence of 46,XY and 46,XX gonadal dysgenesis
phenotype variability in multiple members of an affected
kindred. Horm Res Paediatr 2012;78:119-26. Crossref
23. Josso N, Belville C, di Clemente N, Picard JY. AMH and
AMH receptor defects in persistent Müllerian duct
syndrome. Hum Reprod Update 2005;11:351-6. Crossref
24. Belville C, Van Vlijmen H, Ehrenfels C, et al. Mutations
of the anti-Müllerian hormone gene in patients with
persistent Müllerian duct syndrome: biosynthesis,
secretion, and processing of the abnormal proteins and
analysis using a three-dimensional model. Mol Endocrinol
2004;18:708-21. Crossref
25. Consortium on the Management of Disorders of Sex
Development: Clinical Guidelines for the Management of
Disorders of Sex Development in Childhood. California,
US: Intersex Society of North America; 2006. Available
from: http://www.dsdguidelines.org/files/clinical.pdf.
Accessed Aug 2015.
26. Hersmus R, Stoop H, Turbitt E, et al. SRY mutation
analysis by next generation (deep) sequencing in a cohort
of chromosomal Disorders of Sex Development (DSD)
patients with a mosaic karyotype. BMC Med Genet
2012;13:108. Crossref
27. Kalfa N, Philibert PH, Baskin LS, Sultan C. Hypospadias:
interactions between environment and genetics. Mol Cell
Endocrinol 2011;335:89-95. Crossref