DOI: 10.12809/hkmj144295
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Idiopathic hypertrophic pachymeningitis mimicking prolactinoma with recurrent vision loss
Julie YC Lok, MRCSEd; Nelson KF Yip, FCOphth; Kelvin KL Chong, FCOphth; CL Li, FCOphth; Alvin L Young, FCOphth
Department of Ophthalmology and Visual Sciences, The Chinese University of Hong Kong, Prince of Wales Hospital, Shatin, Hong Kong
Corresponding author: Dr Alvin L Young (youngla@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Idiopathic hypertrophic pachymeningitis is a rare
inflammatory condition with diffuse thickening of
the dura mater, which may cause a compressive effect
or vascular compromise. We report on a 28-year-old
Chinese woman with a history of granulomatous
mastitis 7 years previously and oligomenorrhoea,
headache, blurred vision, and raised prolactin level 2
years previously, that was diagnosed as prolactinoma
and treated conservatively with bromocriptine.
However, she had recurrent bilateral vision loss
when the bromocriptine was stopped. Her symptoms
were resolved by high-dose steroid injection but
remained steroid-dependent. Serial magnetic
resonance imaging scan showed progressive diffuse
thickening of the pachymeningitis with disappearance
of pituitary apoplexy. Lumbar puncture showed
lymphocytosis with no organisms. Open biopsy of
the meninges was performed and histology showed
features of inflammatory infiltrates and vasculitis.
This is an unusual presentation of a rare condition
in this age-group, with co-existing granulomatous
mastitis and chronic otitis media, and is a diagnostic
challenge mimicking pituitary macroadenoma and
meningioma in initial magnetic resonance imaging
scans.
Introduction
Idiopathic hypertrophic pachymeningitis is a rare
inflammatory condition characterised by fibrosis
and thickening of the dura mater. Diagnosis of
idiopathic hypertrophic pachymeningitis requires a
high index of suspicion, as its initial manifestation
could be subtle clinically and radiologically.
Idiopathic hypertrophic pachymeningitis has posed
considerable diagnostic challenges to attending
clinicians, including radiologists, neurologists, and
ophthalmologists because of its highly variable
presentation. Apart from clinical subtlety and
variability, idiopathic hypertrophic pachymeningitis
is also a great imposter because it can mimic other
common and important neurological conditions
such as prolactinoma.
Case report
A 28-year-old Chinese woman with a history
of granulomatous mastitis 7 years previously was noted to have
oligomenorrhoea, nausea, headache, and raised
prolactin level of 1261.0 mIU/L (reference range
[RR], 108.8-557.1 mIU/L) since March 2011. Her
baseline hormone profile was otherwise normal.
Plain magnetic resonance imaging (MRI) scan
showed an enlarged pituitary gland of up to 1.6 cm
(Figs 1a and 1b). At that time she was diagnosed with prolactinoma and treated with bromocriptine; her
prolactin level was well-controlled subsequently.
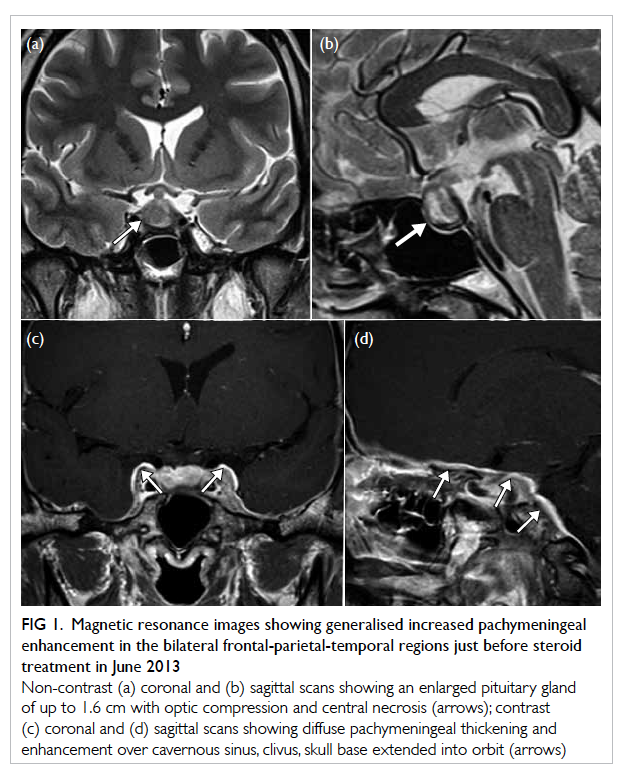
Figure 1. Magnetic resonance images showing generalised increased pachymeningeal enhancement in the bilateral frontal-parietal-temporal regions just before steroid treatment in June 2013
Non-contrast (a) coronal and (b) sagittal scans showing an enlarged pituitary gland of up to 1.6 cm with optic compression and central necrosis (arrows); contrast (c) coronal and (d) sagittal scans showing diffuse pachymeningeal thickening and enhancement over cavernous sinus, clivus, skull base extended into orbit (arrows)
After cessation of bromocriptine at the end
of 2012, she developed periodic headache and
flickering vision loss since May 2013. Her best-corrected
visual acuities were 20/40 and 20/30 in
her right and left eyes, respectively. She developed
red colour desaturation of around 30%. Humphrey
visual field 30-2 test showed superior field loss in
the right eye, while the left eye was full field loss.
Six weeks later her visual acuity decreased to
20/800 in the right eye and 20/400 in the left eye.
She was given dexamethasone and her visual acuity
increased to 20/20 and 20/16 in the right and left
eyes, respectively. Other slit-lamp and fundal
examinations were unremarkable. There was no
papilloedema. There was no involvement of other
cranial nerves. Glasgow Coma Scale Score was 15/15
throughout. Magnetic resonance imaging with
contrast showed increased contrast enhancement
and inflammation over the dura of the sella and
cavernous sinus (Figs 1c and 1d). Her symptoms
resolved and visual function recovered quickly with
high-dose steroid. However, she developed steroid
dependency since repeated attacks developed when
the steroid was stopped.
Serial MRI scan showed progressive diffuse
thickening of the pachymeningitis with disappearance
of pituitary apoplexy, together with chronic otitis
media (Figs 1c and 1d). Lumbar puncture showed 13 cm H2O and lymphocytosis without organisms.
Open biopsy of the meninges was performed and
histology showed features of inflammatory infiltrates
and vasculitis, but was negative for malignancy (Fig
2). Gene rearrangement polymerase chain reaction
assay for immunoglobulin (Ig) heavy chain, T-cell
receptor (TCR)–beta and TCR-gamma all showed
no clonal peak. Polymerase chain reaction for
Mycobacterium tuberculosis and culture for dural
biopsy were also negative; IgG4 was 975 mg/L (RR,
61-1214 mg/L). Complete blood count showed
normal haemoglobin, white blood cell, and platelet
levels. Liver and renal function tests, serum calcium,
creatine kinase, and lactate dehydrogenase levels
were normal. Erythrocyte sedimentation rate was
raised to 113 mm/h (RR, 0-20 mm/h). C-reactive
protein was raised at 67.4 mg/L (reference level,
<9.9 mg/L). In coagulation profile, activated partial
thromboplastin time was slightly prolonged to 40.0
seconds (RR, 28.2-37.4 seconds). Autoimmune
markers, including anti-nuclear antibodies, anti-neutrophil
cytoplasmic antibodies, anti-DNA
immunofluorescence test, anti-extractable nuclear
antigen, anti-cardiolipin antibodies, and rheumatoid
factors are all negative except for the presence of
lupus anticoagulant. Complement C3 was normal
while complement C4 was slightly raised to
0.42 g/L (RR, 0.1-0.4 g/L). Serum vitamin B12 level
was 552 pmol/L (RR, 156-698 pmol/L) and serum
folate level was 29.3 nmol/L (RR, 10.4-42.4 nmol/L);
IgG4 was 975 mg/L. Virology screening—including
human immunodeficiency virus, hepatitis B virus,
and hepatitis C virus—was negative. Serum and
cerebrospinal fluid Venereal Disease Research
Laboratory tests were negative. Chest X-ray was clear
with no features of tuberculosis or sarcoidosis.
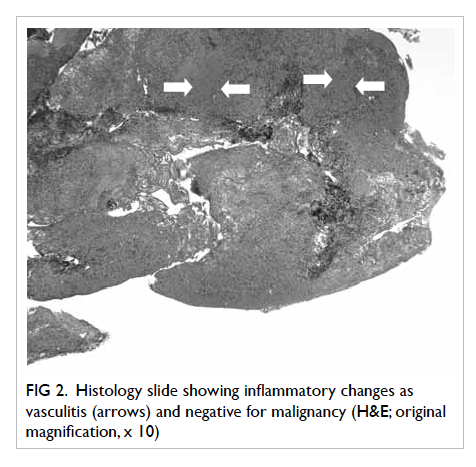
Figure 2. Histology slide showing inflammatory changes as vasculitis (arrows) and negative for malignancy (H&E; original magnification, x 10)
The patient was initially treated with pulse
steroid and was well-controlled by low-dose steroid.
She remained symptom-free 6 months after biopsy.
Discussion
Idiopathic hypertrophic pachymeningitis is a rare
inflammatory condition involving focal and/or
diffuse thickening and fibrosis of the dura mater.
Thickened dura mater with local mass effect may
be pathognomonic of this condition. This pressure
effect may serve as a mechanistic explanation of the
observed neurological defect. As almost every part
of the dura mater can be affected focally and/or
diffusely, there is a highly variable clinical picture.1 2 Diagnosis is almost always made by exclusion of a
large number of aetiologies, for example, infectious
causes such as Lyme disease, syphilis, and M
tuberculosis; inflammatory causes such as Wegener
granulomatosis,3 rheumatoid arthritis, Behçet
disease, and sarcoidosis (which is rare in Chinese
people); and malignancy. In this patient, these tests
were all negative, so the diagnosis of idiopathic
hypertrophic pachymeningitis was made.
Demographically, the median age of patients
with idiopathic hypertrophic pachymeningitis is
58.3 years (standard deviation, 15.8; range, 37-88
years).4 5 6 Only a few paediatric patients have been
reported, with the youngest age being 2 years and
11 months in India.7 Our patient had early-onset
disease. There are few data on ethnicity due to
the rarity of the disease. Idiopathic hypertrophic
pachymeningitis is extremely rare in Chinese people.
The exact aetiopathophysiology of idiopathic
hypertrophic pachymeningitis is not known. It
is believed to be autoimmune in origin.8 Lupus
anticoagulant is a type of autoantibody that binds
to phospholipid and protein, which is commonly
associated with autoimmune diseases such as
systemic lupus erythematous and antiphospholipid
syndrome.9 Our patient’s symptoms and signs did
not fit into any diagnostic category of autoimmune
disease. Only the presence of lupus coagulants
might suggest that idiopathic hypertrophic
pachymeningitis is a form of vasculitis, which
might share some common phenomenon with other
autoimmune diseases, although the true relationship
is controversial.
Clinically, headache is by far the most common
feature and the optic nerve is one of the most common
cranial nerves to be involved, which was the case for
this patient. Multiple cranial nerve involvement,
ataxia, cortical blindness, psychosis, motor function
disturbance, fever, convulsion, and/or loss of
consciousness have all been reported.5 6 Yamada et al2 thought that the inflammatory thickening of the
dura may cause damage to the superior hypophyseal
artery resulting in subarachnoid haemorrhage and
apoplexy in the anterior lobe of the pituitary gland.
The posterior lobe was spared in their patient, who
had a normal hormonal profile, unlike our patient.
The initial enlarged pituitary gland with raised
prolactin was more likely to result from the stalk
effect than from true prolactinoma.
Granulomatous mastitis is a rare idiopathic
chronic benign breast condition, which is believed to
be autoimmune in origin, and mainly affects women
of child-bearing age.10 Granulomatous mastitis is
mainly diagnosed by exclusion of other diagnoses.
To the best of our knowledge, this is the first case of
idiopathic hypertrophic pachymeningitis reported
with the association of granulomatous mastitis,
possibly related to the scarcity of cases affecting
menstruating women.
Idiopathic hypertrophic pachymeningitis
is a rare condition with a highly variable clinical
presentation making accurate and timely diagnosis
difficult. Therefore the attending clinician should
maintain high vigilance in the event of an atypical
presentation of a presumably typical disease. Early
diagnosis and prompt therapeutic intervention such
as high-dose steroid may be the key to preserving
vision as well as life.
References
1. Christakis PG, Machado DG, Fattahi P. Idiopathic
hypertrophic pachymeningitis mimicking neurosarcoidosis.
Clin Neurol Neurosurg 2012;114:176-8. Crossref
2. Yamada SM, Aoki M, Nakane M, Nakayama H. A case of
subarachnoid hemorrhage with pituitary apoplexy caused
by idiopathic hypertrophic pachymeningitis. Neurol Sci
2011;32:455-9. Crossref
3. Yokoseki A, Saji E, Arakawa M, et al. Hypertrophic
pachymeningitis: significance of myeloperoxidase anti-neutrophil
cytoplasmic antibody. Brain 2014;137:520-36. Crossref
4. Yonekawa T, Murai H, Utsuki S, et al. A nationwide
survey of hypertrophic pachymeningitis in Japan. J Neurol
Neurosurg Psychiatry 2014;85:732-9. Crossref
5. Riku S, Kato S. Idiopathic hypertrophic pachymeningitis.
Neuropathology 2003;23:335-44. Crossref
6. Kupersmith MJ, Martin V, Heller G, Shah A, Mitnick HJ.
Idiopathic hypertrophic pachymeningitis. Neurology
2004;62:686-94. Crossref
7. Sharma PK, Saikia B, Sharma R, Gagneja V, Khilnani
P. Pachymeningitis in a young child responded to
antitubercular therapy: a case report. J Child Neurol
2014;29:NP92-5. Crossref
8. Masson C, Boukriche Y, Colombani JM. Inflammatory
hypertrophic cranial pachymeningitis. Presse Med
2001;95:65-70.
9. Favaloro EJ, Wong RC. Antiphospholipid antibody testing
for the antiphospholipid syndrome: a comprehensive
practical review including a synopsis of challenges and
recent guidelines. Pathology 2014;46:481-95. Crossref
10. Poovamma CU, Pais VA, Dolas SC, Prema M, Khandelwal
R, Nisheena R. Idiopathic granulomatous mastitis: a rare
entity with a variable presentation. Breast Dis 2014;34:101-4.