DOI: 10.12809/hkmj134127
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Primitive neuroectodermal adrenal gland tumour
YP Tsang, MB, ChB, MRCS(Ed); Brian HH Lang, MS, FRACS; SC Tam, MB, BS, MRCS(Ed); KP Wong, MB, BS, FRCSEd (Gen)
Division of Endocrine Surgery, Department of Surgery, Queen Mary
Hospital, 102 Pokfulam Road, Hong Kong
Corresponding author: Dr Brian HH Lang (blang@hku.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Ewing’s sarcoma, also called primitive
neuroectodermal tumour of the adrenal gland, is
extremely rare. Only a few cases have been reported
in the literature. We report on a woman with adult-onset
primitive neuroectodermal tumour of the
adrenal gland presenting with progressive flank
pain. Computed tomography confirmed an adrenal
tumour with invasion of the left diaphragm and
kidney. Radical surgery was performed and the
pain completely resolved; histology confirmed the
presence of primitive neuroectodermal tumour, for
which she was given chemotherapy. The clinical
presentation of this condition is non-specific, and
a definitive diagnosis is based on a combination
of histology, as well as immunohistochemical and
cytogenic analysis. According to the literature, these
tumours demonstrate rapid growth and aggressive
behaviour but there are no well-established
guidelines or treatment strategies. Nevertheless,
surgery remains the mainstay of local disease control;
curative surgery can be performed in most patients.
Adjuvant chemoirradiation has been advocated yet
no consensus is available. The prognosis of patients
with primitive neuroectodermal tumours remains
poor.
Introduction
Ewing’s sarcoma (ES), also called primitive
neuroectodermal tumours (PNET), is a rare cancer
of presumed neuroectodermal origin and is mostly
found in children and young adults.1 Since it usually
involves the diaphysis of long bones, adrenal ES/PNET is extremely rare. To our knowledge, only a
handful of cases have been reported in the literature.
Herein we present a patient with adult-onset adrenal
PNET and discuss the diagnostic and management
issues.
Case report
A 37-year-old woman presented to our hospital
in May 2013 with progressive pain over left flank
and abdomen for 1 month. Physical examination
revealed a ballotable mass over the left flank.
Contrast computed tomography (CT) demonstrated
a 12-cm left adrenal mass with infiltration to the left
crus of diaphragm and upper pole of the left kidney
(Fig). On closer examination, the left renal artery was encased by the tumour but there was no evidence
of distant metastases. Hormonal workup of this
adrenal mass suggested that it was a non-functioning
tumour. There was no excess in 24-hour urinary
catecholamines and metanephrines, while 24-hour
urinary free cortisol excretion was 71 (reference range
[RR], 24-140) nmol. The aldosterone-renin ratio
was 9.0 (reference level, <20) ng/dL per ng/(mL•h).
Luteinising hormone, follicle-stimulating hormone,
and testosterone levels were 3.0 (RR, 1.2-103.0) IU/L,
6.8 (1.8-22.5) IU/L, and 0.83 (0.35-2.60) nmol/L,
respectively. The dehydroepiandrosterone level was 4.5 (RR, 2-11) µmol/L. Owing to increasing pain
and the rapidly growing tumour, she underwent
surgery, which revealed a 12-cm adrenal tumour
compressing the aorta and superior mesenteric
artery, and the posterior part of the diaphragm and
left renal artery were invaded by the tumour bulk.
Accordingly, she underwent a left adrenalectomy
together with radical nephrectomy and partial
resection of the diaphragm. No gross tumour was
left behind. Thereafter, she made a good uneventful
recovery and was discharged on the fifth day. Her
back pain resolved completely after surgery.
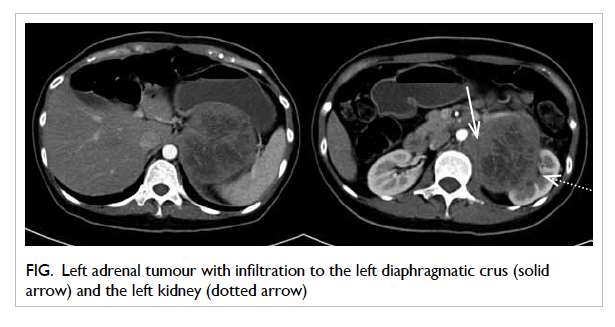
Figure. Left adrenal tumour with infiltration to the left diaphragmatic crus (solid arrow) and the left kidney (dotted arrow)
Histology revealed a monotonous population
of small round tumour cells with hyperchromatic
nuclei, small nucleoli, and minimal cytoplasm
arranged in sheets and cords with occasional
vague rosette formation. Karyorrhexis, mitosis,
and ‘starry sky’ pattern were focally observed.
Immunohistochemically, the tumour cells stained
positively for MIC2 antigen (CD99), vimentin,
and FLI1. By contrast they stained negatively for
cytokeratin, leukocyte common antigen, MPO, WT1,
synaptophysin, S100, desmin, myogenin, CD68, and
CD34. The reverse transcriptase–polymerase chain
reaction confirmed a reciprocal translocation of
t(11;22)(q24;q12) involving the EWSR1 gene on
chromosome 22 and the FLI1 gene on chromosome
11 (ie EWSR1-FLI1 translocation). Overall, these
findings were consistent with ES/PNET.
She received adjuvant chemotherapy
(cyclophosphamide, adriamycin, and vincristine
alternating with ifosfamide and etoposide). The latest
CT at 5 months post-surgery revealed no evidence of
local or distant recurrence.
Discussion
Ewing’s sarcoma/PNET of the adrenal gland is rarely
reported.2 3 Our review of the English literature
revealed only 10 reports describing 16 patients
(Table).3 4 5 6 7 8 9 10 11 12 As in our patient, CD99, a highly sensitive
marker for PNET, was found in all 16 of these
patients. Although not routinely looked for, as in our
patient over 90% of PNET cases exhibit a reciprocal
translocation of t(11;22)(q24;q12) involving the
EWSR1 gene on chromosome 22 and the FLI1 gene
on chromosome 11.1 3 6 7 8 9 11 12 These 16 reported cases
had a median age at diagnosis of 26 (range, 20-74)
years, and the female-to-male ratio was 3:1. The
median tumour size was 10.5 (range, 3-17) cm.
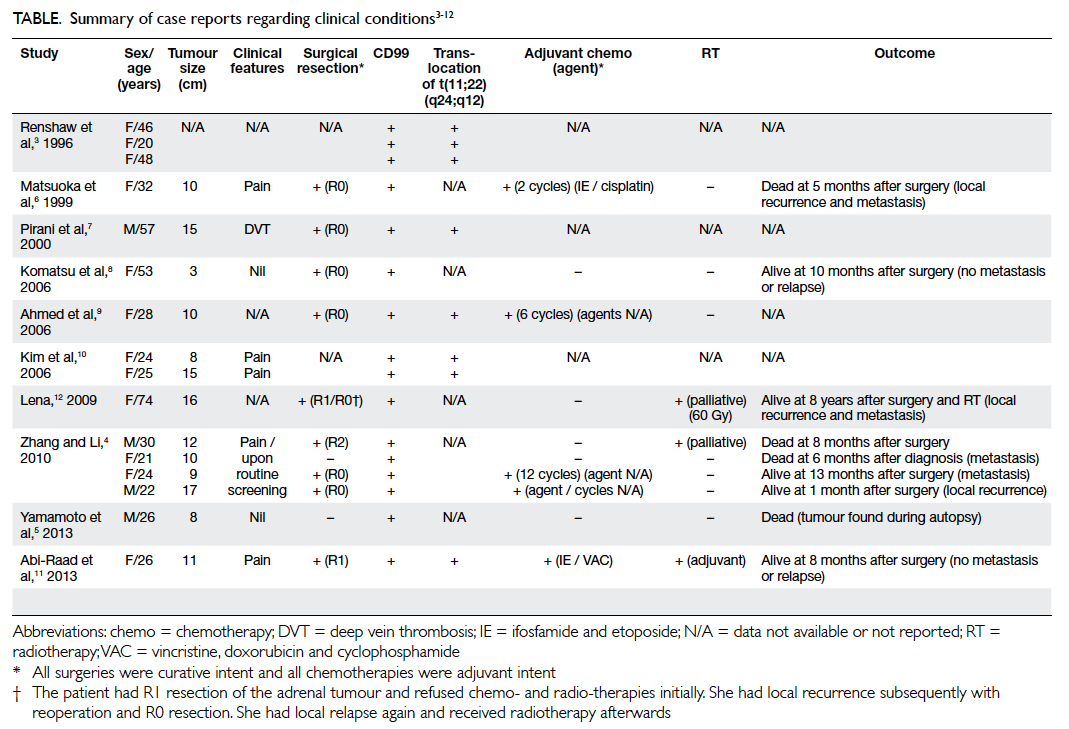
Nine of these patients underwent curative
surgery despite rapid growth and aggressive tumour
behaviour.4 7 8 12 13 Nevertheless, in a few patients
the tumour behaves indolently or has a relatively quiescent period before rapid growth. A case of
incidental PNET at autopsy has been reported in a 26-year-old man who committed suicide by hanging himself; his latent PNET measured 8 cm.5 Similarly, another patient aged 53 years underwent a laparoscopic adrenalectomy for a presumed non-functional incidental adrenocortical adenoma, but turned out to be a 3-cm PNET.8
Diagnostic issues
It is difficult to make a definitive diagnosis of ES/PNET before surgery because currently it is based on a combination of histology, immunohistochemistry, and cytogenetic analysis.4 8 Although needle biopsy is a possible approach to making a definitive diagnosis
without resection, it is rarely performed unless the tumour is considered not resectable or adrenal
metastases are suspected. Since our patient had a potentially resectable tumour with no evidence
of distant metastases, needle biopsy was not considered. Furthermore, she was clearly suffering
pain arising from the tumour and therefore surgical resection was clearly indicated.
Management issues
Due to its rarity, there are no well-established
guidelines or treatment strategies for PNET. Surgical
resection remains the mainstay for local disease
control.4 7 8 Since the tumour is believed to be both
chemo- and radio-sensitive, adjuvant chemotherapy
and radiotherapy have often been advocated for
better local and distant control, but there is no
consensus.13 A standard regimen of chemoirradiation
is not yet established due to the sporadic nature of
cases.13 Most chemotherapy regimens for adult-onset
PNET are akin to those for ES in children as both share the same origin.4 14 Chemotherapeutic agents such as cyclophosphamide, adriamycin, vincristine,
ifosfamide, or etoposide have been used.4 7 8 Recently, it has been noted that chemotherapy might be
effective only for the first few cycles, and that the tumours develop resistance very quickly.4 Adjuvant radiotherapy had been used for local recurrences as well as unresectable or incompletely resected
PNETs.4 12 From our review, adrenalectomies were
performed for 11 patients, while chemotherapy and radiotherapy was also given to six and three of
them, respectively. Follow-up revealed that these tumours were aggressive; three of the 16 patients
died within 1 year of diagnosis or surgery. Regarding the six patients who were still surviving when this
report was submitted, one had distant metastasis and another two had local recurrence. Because of
this high rate of recurrence, intensive follow-up with regular CT scans (every 6 months) has been
advocated even after seemingly curative surgery.12
Conclusion
Herein we report a rare case of adult adrenal PNET. The clinical presentation is often vague and non-specific and a definitive diagnosis depends on a combination of histology, immunohistochemistry, and cytogenetic analysis. Surgical resection remains the mainstay of treatment coupled with adjuvant chemoirradiation. Nevertheless, the prognosis
appears poor.
References
1. Grier HE. The Ewing family of tumors. Ewing’s sarcoma
and primitive neuroectodermal tumors. Pediatr Clin North
Am 1997;44:991-1004. CrossRef
2. Quezado M, Benjamin DR, Tsokos M. EWS/FLI-1 fusion
transcripts in three peripheral primitive neuroectodermal
tumors of the kidney. Hum Pathol 1997;28:767-71. CrossRef
3. Renshaw AA, Perez-Atayde AR, Fletcher JA, Granter SR.
Cytology of typical and atypical Ewing’s sarcoma/PNET.
Am J Clin Pathol 1996;106:620-4.
4. Zhang Y, Li H. Primitive neuroectodermal tumors of
adrenal gland. Jpn J Clin Oncol 2010;40:800-4. CrossRef
5. Yamamoto T, Takasu K, Emoto Y, et al. Latent adrenal
Ewing sarcoma family of tumors: a case report. Leg Med
(Tokyo) 2013;15:96-8. CrossRef
6. Matsuoka Y, Fujii Y, Akashi T, Gosehi N, Kihara K.
Primitive neuroectodermal tumour of the adrenal gland.
BJU Int 1999;83:515-6. CrossRef
7. Pirani JF, Woolums CS, Dishop MK, Herman JR. Primitive
neuroectodermal tumor of the adrenal gland. J Urol
2000;163:1855-6. CrossRef
8. Komatsu S, Watanabe R, Naito M, et al. Primitive
neuroectodermal tumor of the adrenal gland. Int J Urol
2006;13:606-7. CrossRef
9. Ahmed AA, Nava VE, Pham T, et al. Ewing sarcoma family
of tumors in unusual sites: confirmation by rt-PCR. Pediatr
Dev Pathol 2006;9:488-95. CrossRef
10. Kim MS, Kim B, Park CS, et al. Radiologic findings of
peripheral primitive neuroectodermal tumor arising in the
retroperitoneum. AJR Am J Roentgenol 2006;186:1125-32. CrossRef
11. Abi-Raad R, Manetti GJ, Colberg JW, Hornick JL, Shah JG,
Prasad ML. Ewing sarcoma/PNET arising in the adrenal
gland. Pathol Int 2013;63:283-6. CrossRef
12. Lena M. Retroperitoneal primitive neuroectodermal
tumour (PNET). A case report and review of the literature.
Rep Practical Oncol Radiother 2009;14:221-4. CrossRef
13. Malpica A, Moran CA. Primitive neuroectodermal tumor of
the cervix: a clinicopathologic and immunohistochemical
study of two cases. Ann Diagn Pathol 2002;6:281-7. CrossRef
14. Bisogno G, Carli M, Stevens M, et al. Intensive
chemotherapy for children and young adults with
metastatic primitive neuroectodermal tumors of the soft
tissue. Bone Marrow Transplant 2002;30:297-302. CrossRef