DOI: 10.12809/hkmj134044
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
An unusual cause of acromegaly
KY Lock, FHKCP, FHKAM (Medicine); IT Lau, FRCP, FHKCP; CK Yeung, FHKCP, FHKAM (Medicine); CP Chan, FHKCP, FHKAM (Medicine)
Department of Medicine, Tseung Kwan O Hospital, Tseung Kwan O, Hong
Kong
Corresponding author: Dr KY Lock (athenalock@live.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
We report a rare case of acromegaly due to a growth
hormone releasing hormone–secreting bronchial
carcinoid tumour. A 40-year-old man initially
presented with acromegalic features, and was
subsequently found to have a large lung mass in the
right lower zone on chest X-ray. Right lower lobectomy
was performed, and the tumour was confirmed
to be a bronchial carcinoid tumour on histology.
Resection of the tumour led to normalisation of
serum insulin-like growth factor 1 level and growth
hormone responses to an oral glucose tolerance test.
Case report
In September 2010, a 40-year-old man presented
to a general practitioner with multiple skin
tags and acanthosis nigricans. He was noted to
have acromegalic features, including prominent
supraorbital ridge, prognathism, and spade-like
hands. He was referred to Tseung Kwan O Hospital
medical out-patient clinic. In the interim, he visited a
private endocrinologist. Investigations demonstrated
that his serum insulin-like growth factor 1 (IGF-1)
level was markedly elevated, being 719 (reference
range, 101-267) ng/mL, and his serum growth
hormone (GH) levels were not suppressed following
an oral glucose tolerance test (OGTT) with a trough
GH level of 9.9 ng/mL, which confirmed the diagnosis
of acromegaly. Magnetic resonance imaging (MRI)
of the pituitary gland (Fig 1a and b) revealed a bulky
pituitary gland with a 1.3 x 1.0 x 1.1 cm (transverse
x height x anteroposterior dimensions) subtle
roundish area in the central anterior part of the
gland. Transsphenoidal resection of the pituitary
macroadenoma was planned. However, preoperative
chest X-ray (Fig 2a) showed a large mass at the right
lower zone. Thus, the operation was cancelled and
computed tomography (CT) thorax showed a well-defined
lobulated mass (Fig 2b and c), measuring
8.6 x 7.4 x 7.1 cm (lateral x anteroposterior x
craniocaudal dimensions), at the basal region of
the right lower lobe. Fluorodeoxyglucose-positron
emission tomography (FDG-PET) of the whole
body suggested that the mass was consistent with a
primary lung cancer; there were no intrapulmonary
or distant metastases. Right lower lobectomy was
performed by a private cardiothoracic surgeon in
October 2010. Histology confirmed the tumour to
be an atypical bronchial carcinoid. He was first seen by us in November 2012. Evaluation showed that his
IGF-1 level and GH response after having an OGTT had
normalised. Repeat MRI of the pituitary gland 17
months after the lobectomy (Fig 1c and d) showed
that the gland had decreased in size compared with
its earlier size and the previously noted structural
lesion had vanished. In light of the co-existence
of bronchial carcinoid and a history of a pituitary
lesion, multiple endocrine neoplasia type 1 (MEN-1)
syndrome was suspected, but genetic testing could
not detect any mutations. Although growth hormone–releasing hormone (GHRH) level was not available,
the patient most likely suffered from a GHRH-secreting
bronchial carcinoid as suggested by the
presence of a histologically confirmed bronchial
carcinoid tumour, and normalisation of serum IGF-1
level and normal GH response following an OGTT
upon complete removal of his lung tumour.
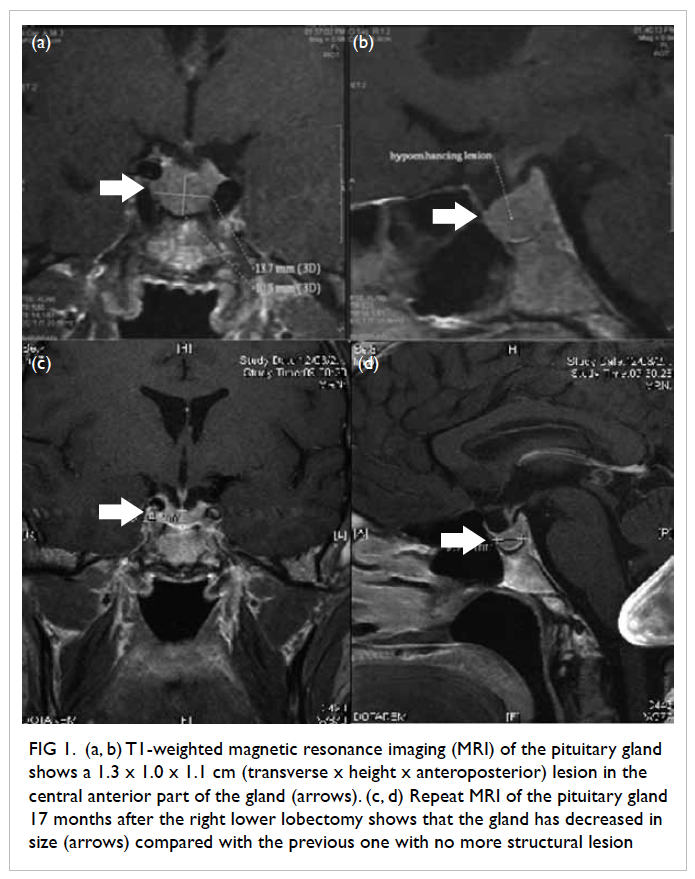
Figure 1. (a, b) T1-weighted magnetic resonance imaging (MRI) of the pituitary gland shows a 1.3 x 1.0 x 1.1 cm (transverse x height x anteroposterior) lesion in the central anterior part of the gland (arrows). (c, d) Repeat MRI of the pituitary gland 17 months after the right lower lobectomy shows that the gland has decreased in size (arrows) compared with the previous one with no more structural lesion
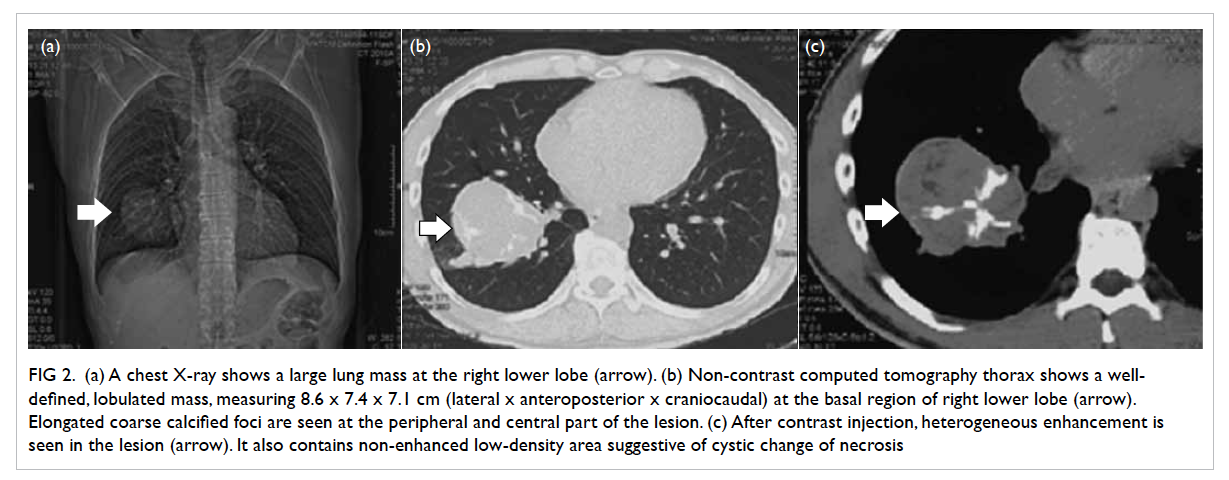
Figure 2. (a) Chest X-ray shows a large lung mass at the right lower lobe (arrow). (b) Non-contrast computed tomography thorax shows a well-defined, lobulated mass, measuring 8.6 x 7.4 x 7.1 cm (lateral x anteroposterior x craniocaudal) at the basal region of right lower lobe (arrow). Elongated coarse calcified foci are seen at the peripheral and central part of the lesion. (c) After contrast injection, heterogeneous enhancement is seen in the lesion (arrow). It also contains non-enhanced low-density area suggestive of cystic change of necrosis
Discussion
Acromegaly is due to sustained and unregulated
hypersecretion of GH. It develops insidiously
and progresses slowly, and typically remains
undiagnosed for about 10 years.1 More than 95%
of cases are caused by autonomous secretion of
GH from anterior pituitary tumours and result in
clonal expansion of somatotrophs. Less than 1% are
due to ectopic GHRH production, with bronchial
carcinoids being the most common cause (70%)
followed by pancreatic islet cell carcinoids.2
Since the majority of bronchial carcinoids arise
in the proximal airways, patients usually present with
pulmonary symptoms,3 including cough, shortness of
breath, wheeze, haemoptysis, chest pain, or recurrent
pneumonia in the same pulmonary segment or lobe
(due to bronchial obstruction). Although many of the tumours express immunoreactive GHRH, most
patients with bronchial carcinoid are not clinically
acromegalic. The first case of a bronchial carcinoid
causing acromegaly was reported in 1958.4 Despite
the large size and central location of his tumour, our
patient did not have any chest symptoms; instead
he came to medical attention because of prominent acromegalic features.
The clinical manifestations of acromegaly
in patients with the ectopic GHRH syndrome are
indistinguishable from those of any GH-secreting
pituitary adenoma.5 Similarly, regardless of the cause,
serum GH and IGF-1 levels are invariably elevated
and GH levels fail to suppress (<1 ng/mL) during
OGTT in all forms of acromegaly.6 No dynamic tests
are helpful in differentiating the causes.7 Among
all, plasma GHRH is the most precise and cost-effective
test for the diagnosis of ectopic GHRH
causing acromegaly. Plasma GHRH levels are
usually elevated in patients with peripheral GHRH-secreting
tumours, and are normal or low in patients
with pituitary acromegaly.8 Regrettably, before the
operation plasma GHRH level was not checked in
our patient as this test was not available in most of
the local hospitals. The presence of positive staining
for GHRH could also provide direct evidence of the
diagnosis.
Bronchial carcinoids are usually picked up
easily on chest X-rays and by CT thorax. Compared
with chest X-rays, CT delineates the extent of the
tumour and its location better as well as the presence
of any mediastinal lymphadenopathy. In our patient,
the bronchial carcinoid was visualised by FDG-PET.
However, FDG-PET yields conflicting results when it
comes to identifying bronchial carcinoids, probably
because of their small size and hypometabolic
nature. In a retrospective review of 16 patients
with surgically resected bronchial carcinoids,
preoperative PET detected only 12 (75%).9 The use
of other PET tracers, such as 11C-L-DOPA and 11C-5-hydroxytryptophan, improves the sensitivity for
imaging neuroendocrine tumours.10 Approximately
80% and 60% of typical and atypical bronchial
carcinoids express somatostatin receptors by
immunohistochemistry, respectively. They may also
be imaged with octreoscan.11 However, specificity is
limited because scintigraphy is positive in many other
tumours, and not all carcinoid tumours that express
somatostatin receptors by immunohistochemistry
test positive with octreoscan.
Pituitary gland MRI is necessary to verify the
presence and size of a pituitary lesion, even when
the diagnosis of ectopic GHRH syndrome has been
established. In contrast to patients with classical
acromegaly, no pituitary tumour but an enlargement
of the sella is detected in the majority of such
patients.12 The first MRI of the pituitary gland in
our patient suggested the presence of an anterior
pituitary macroadenoma, which is unexpected in
patients with GHRH-secreting bronchial carcinoid.
Thus, three other issues need to be considered.
First, the co-existing carcinoid tumour and possible
pituitary adenoma alerted us to the possibility of
MEN-1. Second, the bronchial carcinoid might
have metastasised to the pituitary gland. Third, the acromegaly really was due to the pituitary tumour
producing excessive amounts of GH, and that its
auto-infarction leads to normalisation of IGF-1, a
normal GH response after an OGTT and shrinkage
of the tumour on subsequent MRI. The absence of
an MEN-1 mutation and hyperparathyroidism, and
the resolution of pituitary lesion after lobectomy
make the first possibility unlikely. Although we did
not have any histology from the pituitary, again,
disappearance of the lesion after the lobectomy also
makes the second possibility unlikely. Regarding the
third possibility, it cannot be proved or disproved
in the absence of a plasma GHRH level and tumour
histology. Nevertheless, we have to follow the patient
closely to obtain the final answer.
Surgical resection of the bronchial carcinoids
offers the best chance of cure, the prognosis of
following resection of a typical carcinoid is excellent,
with reported 5-year survival rates of 87% to 100%.
While for atypical carcinoid, 5-year survival of 30%
to 95% has been reported.3 13 Chemotherapy and
radiotherapy are generally not effective. For those
with non-resectable, disseminated tumours; who refuse surgery; or who are unsuitable because of
medical co-morbidities, long-acting somatostatin
analogues provide an effective option to control
symptoms, and according to some studies, may also
slow tumour progression.14
Conclusions
Ectopic GHRH acromegaly is so rare that routine
screening would have a very low yield. Instead,
clinicians should bear this diagnosis in mind,
and search for an extrapituitary source of GH
excess in those with unexpected clinical features
(eg breathlessness, wheeze, or facial flushing), absence of a pituitary tumour on imaging, and the
presence of tumours known to be associated with
extrapituitary acromegaly. Measurement of plasma
GHRH is the most cost-effective means of arriving
at a diagnosis, but is not widely available. Chest
X-ray, CT thorax and abdomen could be performed,
if plasma GHRH testing is not available. A correct
diagnosis is important, as the primary treatment for
extrapituitary acromegaly entails surgical removal
of the underlying tumour. Long-acting somatostatin
analogues might be used to control symptoms, if
resection is incomplete or not feasible.
References
1. Cordero RA, Barkan AL. Current diagnosis of acromegaly.
Rev Endo Metab Discord 2008;9:13-9. CrossRef
2. Sano T, Asa SL, Kovacs K. Growth hormone-releasing
hormone-producing tumors: clinical, biochemical, and
morphological manifestations. Endocr Rev 1988;9:357-73. CrossRef
3. Skuladottir H, Hirsch FR, Hansen HH, Olsen JH.
Pulmonary neuroendocrine tumors: incidence and
prognosis of histological subtypes. A population-based
study in Denmark. Lung Cancer 2002;37:127-35. CrossRef
4. Atmann HW, Schutz W. Uber ein knochenhaltiges
Bronchuskarzinoid [in German]. Beitr Pathol Anat 1958;120:455-73.
5. Agha A, Farrell L, Downey P, Keeling P, Leen E, Sreenan
S. Acromegaly secondary to growth hormone releasing
hormone secretion. Ir J Med Sci 2005;173:215-6. CrossRef
6. Bonadonna S, Doga M, Gola M, Mazziotti G, Giustina
A. Diagnosis and treatment of acromegaly and its
complications: consensus guidelines. J Endocrinol Invest
2008;28(11 Suppl International):43-7.
7. Giustina A, Schettino M, Bodini C, Doga M, Licini M,
Giustina G. Effect of galanin on the growth hormone (GH)
to GH-releasing hormone in acromegaly. Metabolism
1992;41:1291-4. CrossRef
8. Mayo KE. Molecular cloning and expression of a pituitary
receptor for growth hormone-releasing hormone. Mol
Endocrinol 1991;6:1734-44. CrossRef
9. Daniels CE, Lowe VJ, Aubry MC, Allen MS, Jett JR.
The utility of fluorodeoxyglucose positron emission
tomography in the evaluation of carcinoid tumors
presenting as pulmonary nodules. Chest 2007;131:255-60. CrossRef
10. Orlefors H, Sundin A, Garske U, et al. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a
universal imaging technique for neuroendocrine tumors:
comparison with somatostatin receptor scintigraphy
and computed tomography. J Clin Endocrinol Metab
2005;90:3392-400. CrossRef
11. Granberg D, Sundin A, Janson ET, Oberg K, Skogseid B,
Westlin JE. Octreoscan in patients with bronchial carcinoid
tumours. Clin Endocrinol (Oxf) 2003;59:793-9. CrossRef
12. Losa M, Schopohl J, von Werder K. Ectopic secretion of growth hormone-releasing hormone in man. J Endocrinol
Invest 1993;16:69-81. CrossRef
13. Asamura H, Kameya T, Matsuno Y, et al. Neuroendocrine
neoplasms of the lung: a prognostic spectrum. J Clin Oncol
2006;24:70-6. CrossRef
14. Drange MR, Melmed S. Long-acting lanreotide induces
clinical and biochemical remission of acromegaly caused
by disseminated growth hormone-releasing hormone-secreting
carcinoid. J Clin Endocrinol Metab 1998;83:3104-9. CrossRef