Hong Kong Med J 2025;31:Epub 2 Dec 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Successful treatment of adult atypical haemolytic uraemic syndrome with multi-organ involvement: a case report
YK Yam, MB, ChB, MRCP1; Alison LT Ma, FRCPCH, FHKAM (Paediatrics)2; Zoe SY Tsang, MB, ChB, MRCP1; SK Yuen, FRCP, FHKAM (Medicine)1
1 Department of Medicine and Geriatrics, Caritas Medical Centre, Hong Kong SAR, China
2 Department of Paediatrics and Adolescent Medicine, Hong Kong Children’s Hospital, Hong Kong SAR, China
Corresponding author: Dr SK Yuen (yuensk@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Case presentation
A 51-year-old Chinese woman presented with status
epilepticus in May 2024. Four months previously, she
had been admitted for abdominal pain. Computed
tomography (CT) of the abdomen and pelvis at the
time was unremarkable. She had remained well until
she developed upper respiratory tract infection
symptoms, followed by recurrent abdominal
pain and vomiting for 2 days. Her mental status
deteriorated with irritability and mutism, followed
by recurrent generalised tonic-clonic seizures.
Physical examination was unremarkable. She was
intubated and managed in the intensive care unit
with multiple anticonvulsants.
The initial plain CT of the brain was normal,
but follow-up CT revealed new bilateral cerebellar,
thalamic and occipital hypodensities (Fig 1). Blood
tests showed thrombocytopenia (platelet count:
36×109/L, reference: 145-370) and acute kidney
injury (creatinine level: 663 μmol/L, reference: 49-83).
Haemoglobin level dropped to 6.6 g/dL (reference: 11.7-14.9) over the subsequent days with identifiable
schistocytes, raised lactate dehydrogenase level
(2532 U/L, reference: 103-199), raised indirect
bilirubin level (direct-to-total bilirubin: 14:39 μmol/L),
undetectable haptoglobin level (<0.07 g/L, reference:
0.30-2.00), reticulocytosis (133.2×109/L, reference:
20-101; 5.9%) and negative direct antiglobulin
test, suggestive of microangiopathic haemolytic
anaemia. Kidney biopsy confirmed thrombotic
microangiopathy (TMA) with congested glomeruli,
double contours, and capillary thrombi on fibrin
staining (Fig 2).
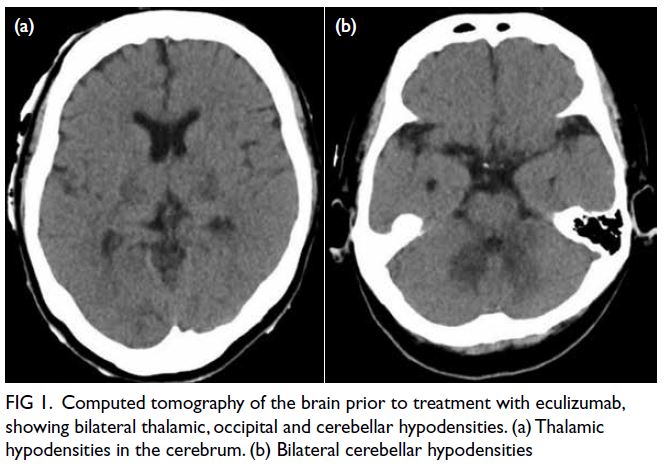
Figure 1. Computed tomography of the brain prior to treatment with eculizumab, showing bilateral thalamic, occipital and cerebellar hypodensities. (a) Thalamic hypodensities in the cerebrum. (b) Bilateral cerebellar hypodensities
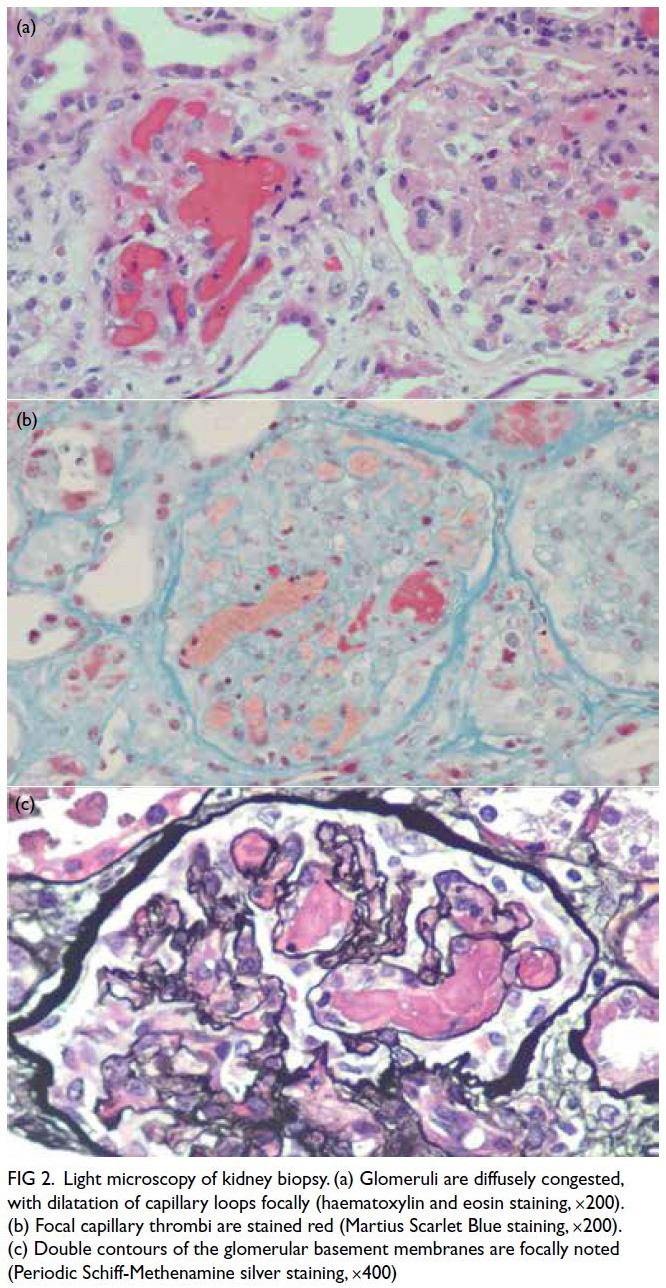
Figure 2. Light microscopy of kidney biopsy. (a) Glomeruli are diffusely congested, with dilatation of capillary loops focally (haematoxylin and eosin staining, ×200). (b) Focal capillary thrombi are stained red (Martius Scarlet Blue staining, ×200). (c) Double contours of the glomerular basement membranes are focally noted (Periodic Schiff-Methenamine silver staining, ×400)
ADAMTS13 activity was normal at 75.4%
(reference: 60.6%-130.6%; <10% signifies severe
deficiency1). Stool culture and polymerase chain
reaction for Shiga toxin genes, urine Streptococcus
pneumoniae soluble polysaccharide antigen,
polymerase chain reaction of nasopharyngeal
swab for common respiratory viruses, as well as
serological tests for human immunodeficiency
virus, cytomegalovirus and Epstein-Barr virus
were all negative. Antinuclear antibodies, anti–double-stranded DNA antibodies, antiphospholipid
antibodies, antibodies to extractable nuclear antigen,
anti–topoisomerase I antibody and antineutrophil
cytoplasmic antibodies were likewise negative.
Complement component 3 and complement
component 4 were normal. Results of homocysteine
and amino acid chromatography excluded cobalamin
C deficiency. Urine tests were negative for pregnancy
and drugs that might induce TMA. There were
no clinical features suggestive of malignancy. The
diagnosis of atypical haemolytic uraemic syndrome
(aHUS) with multisystem (gastrointestinal,
neurological and kidney) involvement, possibly
triggered by upper respiratory tract infection,
was made. The patient was apnoeic and anuric,
necessitating ventilatory and haemodialysis support.
Experience sharing from paediatric nephrology
colleagues facilitated prompt testing for anti–factor
H antibody (anti-FH), complement functional assays
(CH50, AH50, and sC5b9) and genetic analysis for
variants related to aHUS. Urgent application was made through the expert panel of the Hospital
Authority for eculizumab, a complement component
5 (C5) inhibitor. Anti–factor H antibody level
was raised to 11.6 U/mL (reference: <10) but no
pathogenic variants were detected by next-generation
sequencing or multiplex ligation-dependent probe
amplification analysis of the aHUS genetic panel including the following genes: CFH, CFB, CFI, MCP, C3, CFHR1, CFHR3, CFHR5, DGKE and MMACHC
(see online Appendix for the full names of genes).
In view of the likely diagnosis of complement-mediated
HUS, the first dose of eculizumab was
given 7 days after admission. Improvement in
haematological indices and renal recovery were
evident within the first and third week, respectively.
She was extubated 12 days after eculizumab
commencement and had been weaned off
haemodialysis after 4 weeks. Repeat CT of the brain
confirmed resolution of previous abnormalities
(Fig 1). She was seizure-free and ambulatory
with full neurological recovery upon discharge
at 10 weeks post-hospitalisation. Prednisolone
and mycophenolate mofetil were started while
eculizumab was weaned off after 28 weeks of
treatment. Her platelet count, lactate dehydrogenase
level, haptoglobin level and kidney function have
remained normal on serial monitoring.
Discussion
Atypical haemolytic uraemic syndrome is a form of
TMA caused by dysregulation of the complement
pathway.1 The nomenclature of the disease is
evolving.2 In 50% to 60% of patients, either
complement gene variants or anti-FH autoantibodies
result in dysregulation of the alternative pathway.1
Clinical features include microangiopathic
haemolytic anaemia, thrombocytopenia and end-organ
injury, most commonly acute kidney injury.3
Extrarenal manifestations involving neurological,
gastrointestinal, pulmonary and cardiovascular
systems are also seen.4 Half of the affected cases are
preceded by triggers such as infections, medications
and pregnancy.1 The annual incidence of aHUS has
been reported as 0.5 to 2 per million, of whom 41%
to 58% of cases are adults.1 This is contrary to the
traditional belief that it is a childhood condition.1
Under recognition of aHUS might be attributed
to inexperience of the disease and its diagnostic
algorithm.
The diagnosis of aHUS requires urgent
evaluation to exclude other types of TMA, namely
thrombotic thrombocytopenia, Shiga toxin–producing Escherichia coli–haemolytic uraemic
syndrome and different secondary forms.3 It is often
a challenge to differentiate a complement-mediated
TMA with a trigger from a secondary TMA. A full
battery of tests is required to thoroughly exclude
alternative diagnoses once TMA is recognised,5 as in
our case. Complement studies, anti-FH antibody titre
and genetic analysis are essential in the diagnostic
pathway.5 A normal complement component 3
level in our patient was not surprising since it is
reported low in only about 50% of cases.3 Genetic
screening is particularly important as it correlates
with the response to treatment, risk of relapse, and prognosis.3 Nevertheless a negative genetic test does
not exclude the diagnosis of complement-mediated
TMA since about 40% of patients have no variants
identified.5
The outcome of aHUS has been historically
poor prior to the development of complement
inhibitors. Eculizumab and ravulizumab are anti-C5
monoclonal antibodies that block the terminal
complement pathway.5 Eculizumab has been shown
to prolong the 5-year end-stage kidney disease-free
survival of aHUS patients from 39.5% to 85.5%.5 In
Hong Kong, eculizumab or ravulizumab may be
publicly funded for indicated patients, but only with
prior approval from a local expert panel.6 Physicians
should take note of the available resources, including
the diagnostic and management pathways, and the
funding procedure in relation to aHUS to facilitate
appropriate patient care.
This case illustrates the effect response of an
adult patient with complement-mediated aHUS to
C5 inhibitors, highlighting the importance of timely
recognition, evaluation and management of this rare
condition.
Author contributions
Concept or design: YK Yam, SK Yuen.
Acquisition of data: YK Yam.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: YK Yam.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: YK Yam.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: YK Yam.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Acknowledgement
The authors acknowledge the invaluable contribution of the
Intensive Care Unit and the Department of Pathology at the
Caritas Medical Centre in the care of this patient.
Declaration
Preliminary results of the case have been presented as poster
presentation at the 5th International Congress of Chinese
Nephrologists cum Hong Kong Society of Nephrology Annual Scientific Meeting 2024, Hong Kong, 13-15 December 2024.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of
Helsinki. Informed consent for all treatment and procedures
was obtained from the patient or her next-of-kin. Verbal
consent for publication was obtained from the patient.
Supplementary material
The supplementary material was provided by the authors and
some information may not have been peer reviewed. Any
opinions or recommendations discussed are solely those of the
author(s) and are not endorsed by the Hong Kong Academy
of Medicine and the Hong Kong Medical Association.
The Hong Kong Academy of Medicine and the Hong Kong
Medical Association disclaim all liability and responsibility
arising from any reliance placed on the content.
References
1. Donadelli R, Sinha A, Bagga A, Noris M, Remuzzi G. HUS
and TTP: traversing the disease and the age spectrum.
Semin Nephrol 2023;43:151436. Crossref
2. Nester CM, Feldman DL, Burwick R, et al. An expert
discussion on the atypical hemolytic uremic syndrome
nomenclature—identifying a road map to precision: a
report of a National Kidney Foundation Working Group.
Kidney Int 2024;106:326-36. Crossref
3. Goodship TH, Cook HT, Fakhouri F, et al. Atypical
hemolytic uremic syndrome and C3 glomerulopathy:
conclusions from a “Kidney Disease: Improving Global
Outcomes” (KDIGO) Controversies Conference. Kidney
Int 2017;91:539-51. Crossref
4. Schaefer F, Ardissino G, Ariceta G, et al. Clinical and
genetic predictors of atypical hemolytic uremic syndrome
phenotype and outcome. Kidney Int 2018;94:408-18. Crossref
5. Kavanagh D, Ardissino G, Brocklebank V, et al. Outcomes
from the International Society of Nephrology Hemolytic
Uremic Syndromes International Forum. Kidney Int
2024;106:1038-50. Crossref
6. Hong Kong SAR Government. LCQ16: Support for patients
with rare diseases [press release]. 12 Jun 2024. Available
from: https://www.info.gov.hk/gia/general/202406/12/P2024061200662.htm. Accessed 18 Nov 2025.