Hong Kong Med J 2016 Dec;22(6):526–33 | Epub 29 Jul 2016
DOI: 10.12809/hkmj154750
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Silver-Russell syndrome in Hong Kong
HM Luk, MB, BS, FHKAM (Paediatrics)1#;
KS Yeung, BSc, MPhil2#;
WL Wong, BSc, MPhil2;
Brian HY Chung, FHKAM (Paediatrics), FCCMG (Clinical Genetics)2;
Tony MF Tong, MPhil, MSc1;
Ivan FM Lo, MB, ChB, FHKAM (Paediatrics)1
1 Clinical Genetic Service, Department of Health, 3/F, Cheung Sha Wan
Jockey Club Clinic, 2 Kwong Lee Road, Sham Shui Po, Hong Kong
2 Department of Paediatrics and Adolescent Medicine, Queen Mary
Hospital, The University of Hong Kong, Pokfulam, Hong Kong
# Co-first author
 Full
paper in PDF
Full
paper in PDF
Corresponding author: Dr Ivan FM Lo (con_cg@dh.gov.hk)
Full
paper in PDF
Abstract
Objectives: To examine the molecular pathogenetic
mechanisms, (epi)genotype-phenotype
correlation, and the performance of the three clinical
scoring systems—namely Netchine et al, Bartholdi
et al, and Birmingham scores—for patients
with Silver-Russell syndrome in Hong Kong.
Methods: This retrospective case series was
conducted at two tertiary genetic clinics, the Clinical
Genetic Service, Department of Health, and clinical genetic clinic in Queen
Mary Hospital in Hong Kong. All records of patients
with suspected Silver-Russell syndrome under the
care of the two genetic clinics between January
2010 and September 2015 were retrieved from the
computer database.
Results: Of the 28 live-birth patients with Silver-Russell syndrome, 35.7% had H19 loss of DNA
methylation, 21.4% had maternal uniparental
disomy of chromosome 7, 3.6% had mosaic maternal uniparental
disomy of chromosome 11, and the remaining 39.3% were Silver-Russell syndrome of unexplained molecular origin.
No significant correlation between (epi)genotype
and phenotype could be identified between H19
loss of DNA methylation and maternal uniparental
disomy of chromosome 7. Comparison of molecularly confirmed
patients and patients with Silver-Russell syndrome
of unexplained origin revealed that postnatal
microcephaly and café-au-lait spots were more
common in the latter group, and body and limb
asymmetry was more common in the former group.
Performance analysis showed the Netchine et al and
Birmingham scoring systems had similar sensitivity
in identifying Hong Kong Chinese subjects with
Silver-Russell syndrome.
Conclusion: This is the first territory-wide study of
Silver-Russell syndrome in Hong Kong. The clinical
features and the spectrum of underlying epigenetic
defects were comparable to those reported in
western populations.
New knowledge added by this study
- The epigenetic defects of Silver-Russell syndrome (SRS) in Hong Kong Chinese patients are comparable to those reported in western populations.
- No epigenotype-phenotype correlation was demonstrated among SRS patients in this study.
- All suspected SRS patients should be referred to a genetic clinic for assessment.
- A new diagnostic algorithm has been proposed for Chinese patients with SRS.
Introduction
Silver-Russell syndrome (SRS) [OMIM 180860] is a
clinically and genetically heterogeneous congenital
imprinting disorder. It was first described in 1953
by Dr Henry Silver and his colleagues, who reported
two children with short stature and congenital
hemihypertrophy.1 In the following year, Dr
Alexander Russell reported five similar cases with
intrauterine dwarfism and craniofacial dysostosis.2
The term SRS has been used since 1970 to describe
a constellation of features with intrauterine growth
retardation without postnatal catch-up, distinct
facial characteristics, relative macrocephaly, body
asymmetry, and/or fifth finger clinodactyly.3 4 The prevalence of SRS was estimated to be 1 in 100 000,5
but was probably underestimated due to the diverse
and variable clinical manifestations. The majority of
SRS cases are sporadic, although occasional familial
cases have been reported.
Two major molecular mechanisms have been
implicated in SRS—maternal uniparental disomy
of chromosome 7 (mUPD7)6 and loss of DNA
methylation (LOM) of the imprinting control region
1 (ICR1) on the paternal allele of chromosome
11p15 region that regulates the IGF2/H19 locus.6 7 8 9
According to the studies, LOM of ICR1 and
mUPD7 roughly account for 45% to 50% and 5% to
10% of SRS cases, respectively.6 7 8 9 Rare cytogenetic
rearrangements have also been reported in 1% to 2%
of cases.4 10 11 There remain 30% to 40% of SRS cases
in which the molecular mechanisms remain elusive,
however.
Owing to the wide spectrum of clinical
presentations of SRS, there is considerable clinical
overlap with other growth retardation syndromes.
At present there is no consensus for the diagnostic
criteria, so diagnosing SRS is challenging. Several
scoring systems have been proposed to facilitate
clinical diagnosis and to guide genetic testing.7 11 12 13 14
Based on the prevalence of different molecular
mechanisms, methylation study of the 11p15 region
is the recommended first-tier investigation for
patients with suspected SRS, and mUPD7 analysis is
the second tier.14
The comprehensive clinical spectrum and
molecular study of SRS have not been reported in
the Chinese population. Therefore, a retrospective
review that aimed to summarise the clinical and
genetic findings of all SRS patients in Hong Kong
was conducted. The sensitivity and specificity of
different scoring systems7 11 12 13 14 in identifying Hong Kong Chinese SRS patients have also been studied.
Methods
Patients
The Clinical Genetic Service (CGS), Department of Health and the clinical genetic clinic
at Queen Mary Hospital (QMH), The University
of Hong Kong, are the only two tertiary genetic
referral centres that provide comprehensive genetic
counselling, and diagnostic and laboratory service
for the Hong Kong population. Patients with a
clinical suspicion of growth failure due to genetic
causes or possibly SRS were referred for assessment
and genetic testing.
In this review, all records of patients
with suspected SRS seen at the CGS or clinical
genetic clinic of QMH between January 2010 and
September 2015 were retrieved from the computer
database system using the key words of “Silver
Russell syndrome” and “failure to thrive and growth
retardation”. The clinical and laboratory data of these
patients were retrospectively analysed. Patients with
alternative diagnoses after assessment and genetic
investigation were excluded. This study was done in accordance with the principles outlined in the Declaration of Helsinki.
Clinical diagnostic criteria for Silver-Russell
syndrome in this study
Currently, there is no universal consensus on the
diagnostic criteria of SRS, but the Hitchins et al’s
criteria15 are the most commonly used clinically.
The diagnosis of SRS in this study was made when
a patient fulfilled three major, or two major and two
minor criteria.
Major criteria included (1) intrauterine
growth retardation/small for gestational age (<10th
percentile); (2) postnatal growth with height/length
<3rd percentile; (3) normal head circumference
(3rd-97th percentile); and (4) limb, body, and/or facial
asymmetry.
Minor criteria included (1) short arm span
with normal upper-to-lower segment ratio; (2) fifth
finger clinodactyly; (3) triangular facies; and (4) frontal
bossing/prominent forehead.
Epimutation in imprinting control region 1
Investigation of the methylation status and copy
number change of the H19 differentially methylated
region (H19 DMR) and KvDMR1 at chromosome
11p15 region was done with methylation specific–multiplex ligation-dependent probe amplification
(MS-MLPA) method, using SALSA MLPA ME030-B1 BWS/RSS kit (MRC-Holland, Amsterdam,
The Netherlands). Following the manufacturer’s
instructions, approximately 100 ng genomic DNA
was first denatured and hybridised overnight with
the probe mixture supplied with the kit. The samples
were then split into two portions, treated either with
ligase alone or with ligase and HhaI. Polymerase chain
reactions (PCR) were then performed with
the reagents and primers supplied in the kit. The PCR
products were separated by capillary electrophoresis
(model 3130xl; Applied Biosystems, Foster City
[CA], US). The electropherograms were analysed
using GeneScan software (Applied Biosystems,
Foster City [CA], US), and the relative peak area was
calculated using the Coffalyser version 9.4 software
(MRC-Holland, Amsterdam, The Netherlands).
Analysis of maternal uniparental disomy of chromosome 7
We studied mUPD7 with eight polymorphic
microsatellite markers, three on 7p and five on 7q
(D7S531, D7S507, D7S2552, D7S2429, D7S2504,
D7S500, D7S2442, and D7S2465), using a standard
protocol. Haplotype analysis was then performed. A
diagnosis of mUPD7 required evidence of exclusive
maternal inheritance at two or more informative markers.
Data analysis and (epi)genotype-phenotype correlation
Epidemiological data, physical characteristics,
growth records, and molecular findings were
then collected for analysis. Clinical photographs
were taken during consultation (Fig 1). In order to
delineate the (epi)genotype-phenotype correlation,
we divided the patients according to their
(epi)genotype, namely H19 LOM, mUPD7, mosaic
maternal uniparental disomy of chromosome 11 (mUPD11), or SRS of unexplained origin. The SRS of
unexplained origin was defined as negative for 11p15
region epimutation and mUPD7 study. For statistical
calculation, Student’s t test was used for continuous
variables and Fisher’s exact test for categorical
variables. Two-tailed P values were also computed.
Differences were considered to be statistically
significant when P≤0.05.
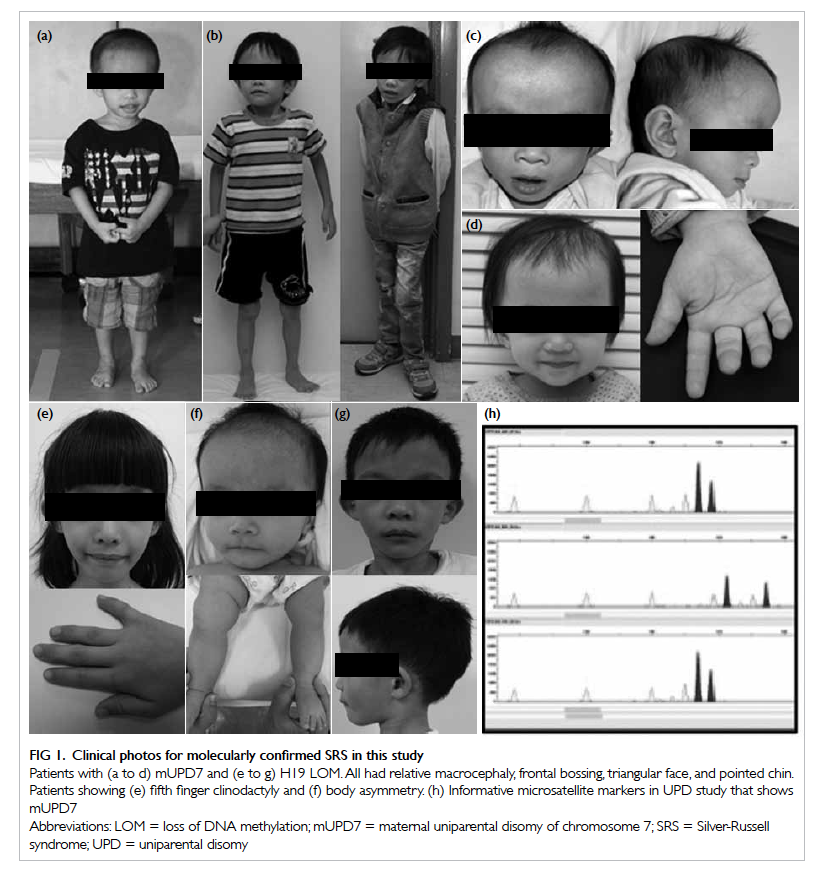
Figure 1. Clinical photos for molecularly confirmed SRS in this study
Patients with (a to d) mUPD7 and (e to g) H19 LOM. All had relative macrocephaly, frontal bossing, triangular face, and pointed chin. Patients showing (e) fifth finger clinodactyly and (f) body asymmetry. (h) Informative microsatellite markers in UPD study that shows mUPD7
Clinical score
Three clinical scoring systems were applied to all
patients referred with suspected SRS and included
Netchine et al score,7 Bartholdi et al score,12 and the Birmingham score.14 An overview of the three SRS
scoring systems is summarised in Table 1. Using the
Hitchins et al’s criteria15 as standard in this study,
the sensitivity and specificity of these three scoring
systems in identifying SRS were compared.
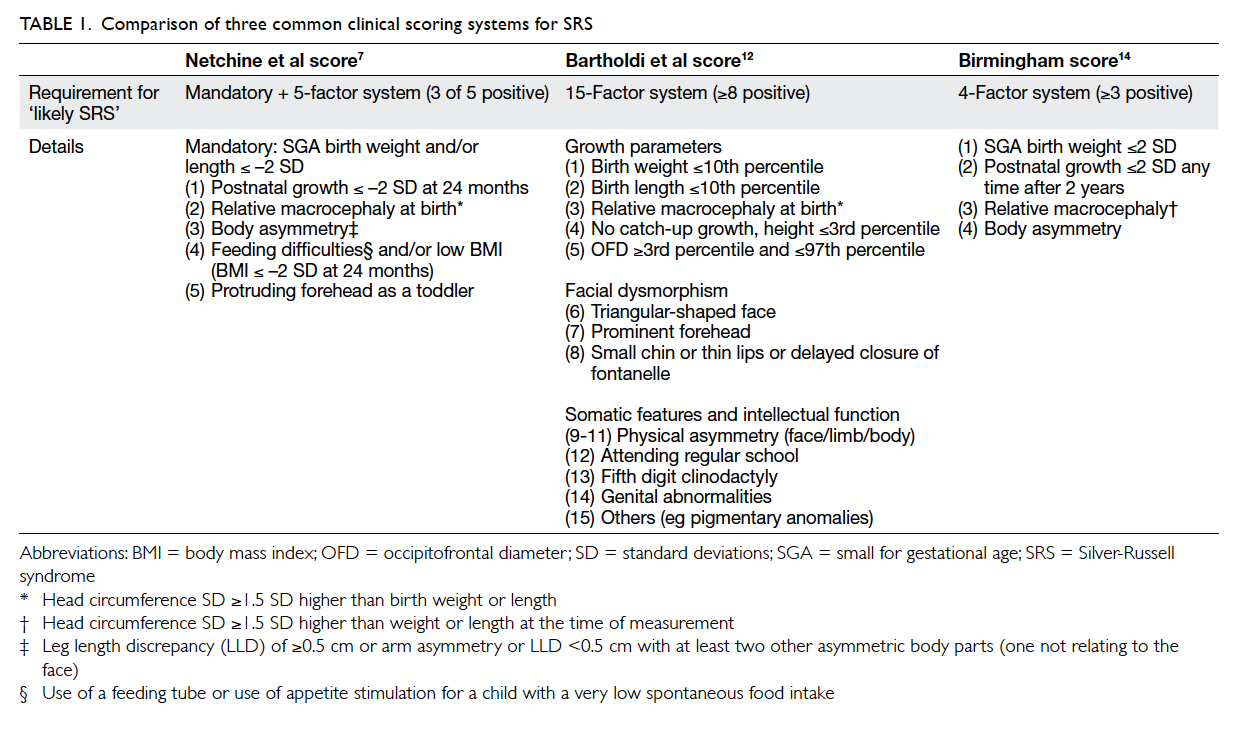
Table 1. Comparison of three common clinical scoring systems for SRS
Results
During the study period, 83 patients with suspected
SRS were referred to both genetic clinics. After
clinical assessment and investigations, 54 patients
had an alternative diagnosis. The remaining 29
patients were clinically diagnosed with SRS using
the Hitchins et al’s criteria.15 All were Chinese. One
was a prenatal case with maternal H19 duplication.
Since termination of pregnancy was performed at 23
weeks of gestation, it was excluded for downstream
analysis. For the remaining 28 SRS patients, their
age at the end of the study (September 2015) ranged
from 2 years to 22 years 9 months, with a median
of 9 years 4 months. The male-to-female ratio was
9:5. Sequential MS-MLPA study on chromosome
11p15 region and mUPD7 study were performed
on all SRS patients. Among the 28 live-birth SRS
patients, 35.7% (n=10) had H19 LOM, 21.4% (n=6)
had mUPD7, 3.6% (n=1) had mosaic mUPD11,
and 39.3% (n=11) were of SRS of unexplained
origin. The clinical features of the SRS cohort are
summarised in Table 2. The clinical features of some
molecularly confirmed SRS patients in this study and
one illustrative microsatellite electropherogram in
mUPD7 analysis are shown in Figure 1.
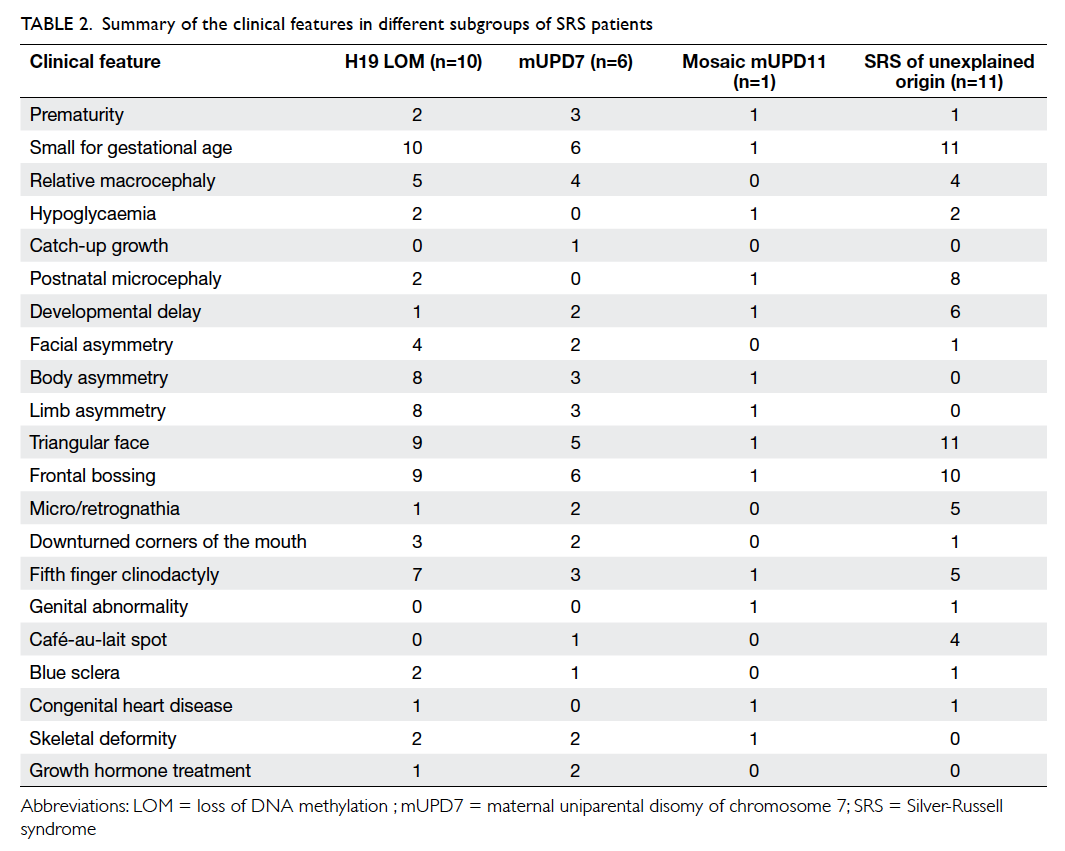
Table 2. Summary of the clinical features in different subgroups of SRS patients
In order to study the (epi)genotype-phenotype
correlation among the H19 LOM and mUPD7
groups, the clinical features were compared.
There was no significant difference among the
two groups (data not shown). When comparing
the 28 molecularly confirmed SRS and 54 SRS of
unexplained origin patients, postnatal microcephaly
(P=0.01) and café-au-lait spots (P=0.05) were more
common among SRS of unexplained origin, while
body asymmetry (P<0.01) and limb asymmetry
(P<0.01) were more common among the molecularly
confirmed group.
The performance of the three clinical scoring
systems namely Netchine et al score,7 Bartholdi et
al score,12 and Birmingham score14 in identifying SRS in our cohort was compared. The proportion
of molecularly confirmed cases in those ‘likely SRS’
and ‘unlikely SRS’ based on the scoring system are
summarised in Table 3. The sensitivity and specificity
among different scoring systems for identifying SRS
are summarised in Table 4.
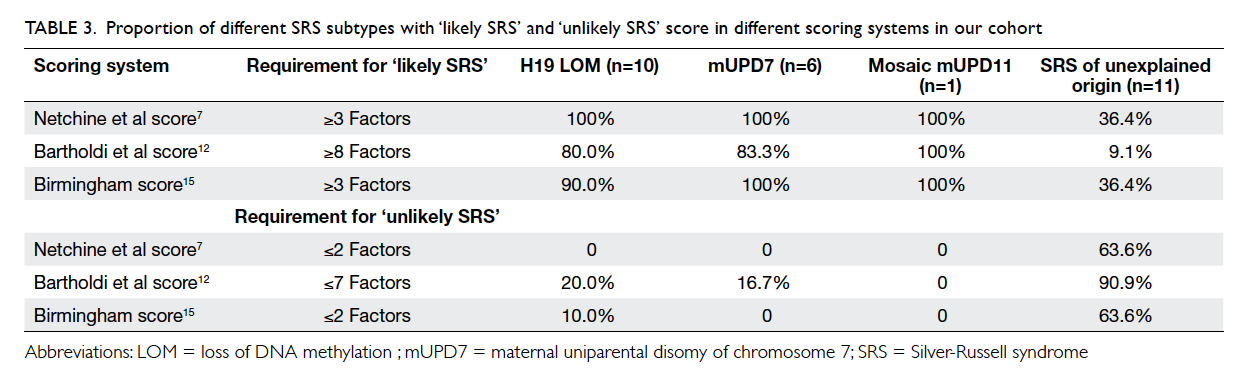
Table 3. Proportion of different SRS subtypes with ‘likely SRS’ and ‘unlikely SRS’ score in different scoring systems in our cohort
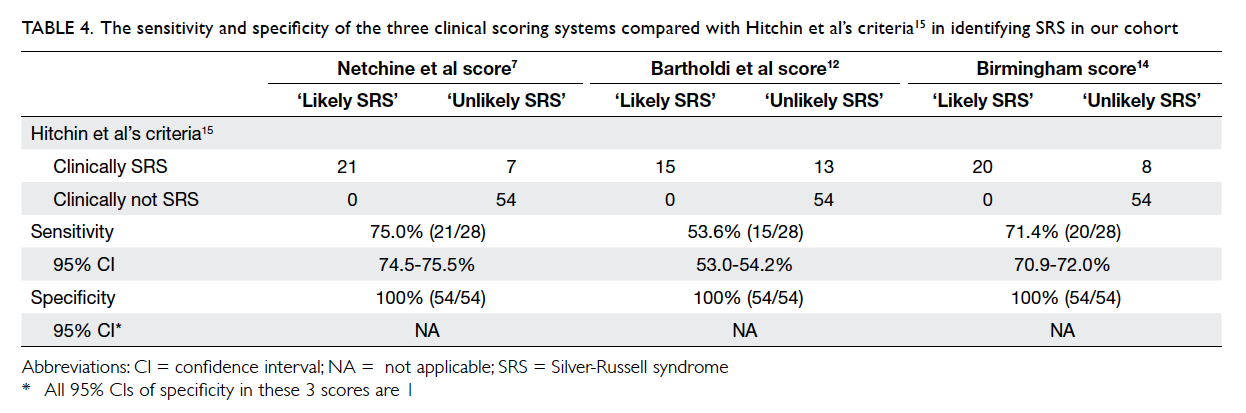
Table 4. The sensitivity and specificity of the three clinical scoring systems compared with Hitchin et al’s criteria15 in identifying SRS in our cohort
Discussion
Silver-Russell syndrome is a clinically and
genetically heterogeneous disorder. This is the first
comprehensive clinical and epigenetic study of SRS
in Hong Kong. With sequential 11p15 epimutation
analysis and mUPD7 study of SRS patients in this
cohort, molecular confirmation was achieved in
60.7% of cases; H19 LOM and mUPD7 accounted for
35.7% and 21.4% of the cases, respectively. Although
the proportion of H19 LOM–related SRS cases was
similar to the western and Japanese populations,6 7 8 9 16 the proportion of mUPD7 in our cohort was
significantly higher. Nonetheless, due to the small
sample size, this observation might not reflect the
true ethnic-specific epigenetic alteration in the
Chinese population. Further studies are necessary to
confirm this difference.
In previous studies of (epi)genotype-phenotype
correlation4 7 12 17 18 19 20 in SRS, patients with mUPD7
had a milder phenotype but were more likely to
have developmental delay. On the contrary, patients
with H19 LOM appeared to have more typical SRS
features such as characteristic facial profile and
body asymmetry. Such a correlation could not be
demonstrated in our cohort. When comparing the
molecularly confirmed and SRS of unexplained
origin groups, postnatal microcephaly and café-au-lait spots were more common in the group of SRS of
unexplained origin, while body/limb asymmetry was
more common in the molecularly confirmed group.
This observation has also been reported in Japanese
SRS patients.16 This might be due to the greater
clinical and genetic heterogeneity in the molecularly
negative SRS.
Although SRS has been extensively studied,
there remains no universal consensus on the
clinical diagnostic criteria. Hitchins et al’s criteria15
are currently the most commonly used. In
order to facilitate the clinical diagnosis, several
additional scoring systems have been proposed
which include the Netchine et al,7 Bartholdi et al,12 and Birmingham scores.14 Each of them has its
advantages and limitations. The major caveats of
those scoring systems include relative subjectivity
of clinical signs, and time-dependent and evolving
clinical features. The heterogeneity of clinical
manifestations also limits their application. In order
to validate these scoring systems, several studies
have been performed to evaluate their accuracy in
predicting the molecular genetic testing result.14 21 We also evaluated the performance of these three
scoring systems in this Chinese cohort. All three
scoring systems are 100% specific in diagnosing
SRS, but the sensitivity for Netchine et al score,7
Bartholdi et al score,12 and Birmingham score14 is 75%, 53.6%, and 71.4%, respectively when compared with
Hitchins et al’s criteria.15 This suggests that Hitchins
et al’s criteria15 remain the most sensitive diagnostic
criteria for SRS when used clinically.
The management of SRS is challenging and
requires multidisciplinary input. Growth hormone
(GH) treatment is the current recommended therapy
for children with small for gestational age without
spontaneous catch-up growth and those with GH
deficiency. In SRS, abnormalities in spontaneous GH
secretion and subnormal responses to provocative
GH stimulation have been well reported.20 The
proposed mechanism is dysregulation of the growth
factors and its major binding protein,4 particularly
in the H19 LOM group. Besides, SRS patients are
expected to have poor catch-up growth. Nonetheless,
GH therapy is not a universal standard treatment for
SRS. In Hong Kong, the indications for GH therapy
under Hospital Authority guidelines do not include
SRS22 without GH response abnormalities. In our
cohort, only three patients who had a suboptimal
GH provocative stimulation test are currently
receiving GH treatment. The long-term outcome is
not yet known.
Although tissue-specific epigenetic
manifestation has been reported in SRS,23 mosaic
genetic or epigenetic alteration is uncommon.24
We have one patient with mUPD11 confirmed by
molecular testing with peripheral blood and buccal
swab samples. Mosaicism should be considered
when a patient has typical SRS phenotype but
negative routine testing. Testing of other tissue
should be pursued so as to provide an accurate
molecular diagnosis that can guide subsequent
genetic counselling and clinical management.
Finally, upon review of the literature, it is well known
that gain of function of the CDKN1C gene25
and maternal UPD14 (Temple syndrome)26 27 can result in a phenotype mimicking SRS. There are also
other syndromic growth retardation disorders with
many overlapping clinical features with those of SRS,
such as mulibrey nanism and 3-M syndrome.28 29 Therefore, with the latest understanding of the
molecular pathogenetic mechanisms of SRS,
together with evidence21 30 31 and results from this
study, we propose the diagnostic algorithm for
Chinese SRS patients as depicted in Figure 2. All
clinically suspected SRS patients should be assessed
by a clinical geneticist. Although the Netchine et
al score,7 Bartholdi et al score,12 and Birmingham score14 are highly specific, they are less sensitive
than the Hitchins et al’s criteria15 for diagnosing
SRS in our Chinese cohort. Therefore, the Hitchins
et al’s criteria15 should be used clinically to classify
those suspected SRS patients into ‘likely’ or ‘unlikely’
SRS. For those ‘likely SRS’ patients, sequential 11p15
region methylation study and mUPD7 analysis should
be performed because 11p15 region epigenetic
alteration is more prevalent than mUPD7 in SRS. For
those molecularly unconfirmed SRS, further testing
for other SRS-like syndromes including Temple
syndrome or CDKN1C-related disorder should be
pursued if indicated.
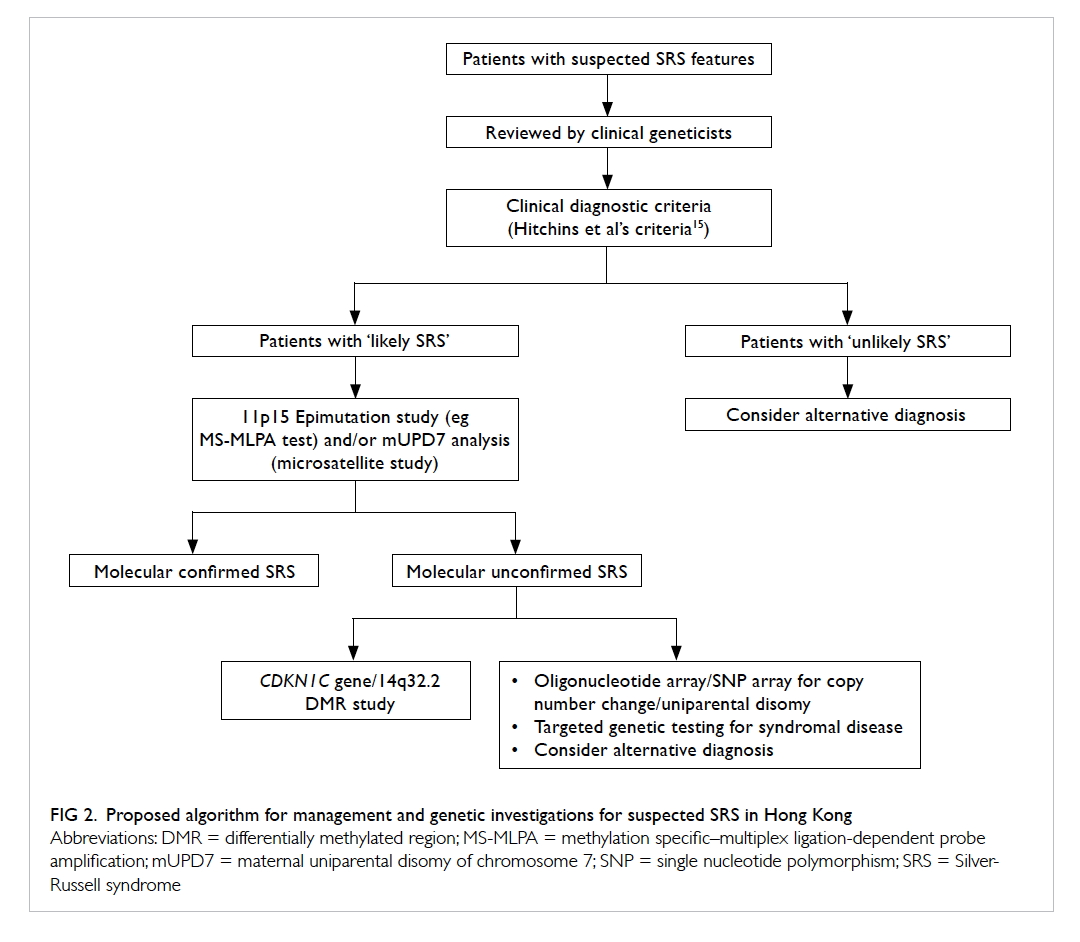
Figure 2. Proposed algorithm for management and genetic investigations for suspected SRS in Hong Kong
Conclusion
This 5-year review is the first territory-wide study
of Chinese SRS patients in Hong Kong. It showed
that the clinical features and underlying epigenetic
mechanisms of Chinese SRS are similar to those
of other western populations. Early diagnosis and
multidisciplinary management are important for
managing SRS patients. Vigilant clinical suspicion
with confirmation by molecular testing is essential.
Based on the current evidence and performance
evaluation of different clinical scoring systems, a
comprehensive diagnostic algorithm is proposed. We
hope that with an increase in understanding of the
underlying pathophysiology and the (epi)genotype-phenotype
correlation in Chinese SRS patients, the
quality of medical care will be greatly improved in
the near future.
Acknowledgements
We thank all the paediatricians and physicians who
have referred their SRS patients to our service and
QMH. We are also grateful to all the laboratory
staff in CGS for their technical support. This work
in HKU is supported by HKU small project funding
and The Society for the Relief of Disabled Children
in Hong Kong.
Declaration
All authors have disclosed no conflicts of interest.
References
1. Silver HK, Kiyasu W, George J, Deamer WC. Syndrome
of congenital hemihypertrophy, shortness of stature, and
elevated urinary gonadotropins. Pediatrics 1953;12:368-76.
2. Russell A. A syndrome of intra-uterine dwarfism
recognizable at birth with cranio-facial dysostosis,
disproportionately short arms, and other anomalies (5
examples). Proc R Soc Med 1954;47:1040-4.
3. Wollmann HA, Kirchner T, Enders H, Preece MA, Ranke
MB. Growth and symptoms in Silver-Russell syndrome:
review on the basis of 386 patients. Eur J Pediatr
1995;154:958-68. Crossref
4. Wakeling EL, Amero SA, Alders M, et al. Epigenotype-phenotype
correlations in Silver-Russell syndrome. J Med
Genet 2010;47:760-8. Crossref
5. Christoforidis A, Maniadaki I, Stanhope R. Managing
children with Russell-Silver syndrome: more than just
growth hormone treatment? J Pediatr Endocrinol Metab
2005;18:651-2. Crossref
6. Kotzot D, Schmitt S, Bernasconi F, et al. Uniparental
disomy 7 in Silver-Russell syndrome and primordial
growth retardation. Hum Mol Genet 1995;4:583-7. Crossref
7. Netchine I, Rossignol S, Dufourg MN, et al. 11p15
Imprinting center region 1 loss of methylation is a common
and specific cause of typical Russell-Silver syndrome:
clinical scoring system and epigenetic-phenotypic
correlations. J Clin Endocrinol Metab 2007;92:3148-54. Crossref
8. Gicquel C, Rossignol S, Cabrol S, et al. Epimutation of the
telomeric imprinting center region on chromosome 11p15
in Silver-Russell syndrome. Nat Genet 2005;37:1003-7. Crossref
9. Schönherr N, Meyer E, Eggermann K, Ranke MB,
Wollmann HA, Eggermann T. (Epi)mutations in 11p15
significantly contribute to Silver-Russell syndrome: but are
they generally involved in growth retardation? Eur J Med
Genet 2006;49:414-8. Crossref
10. Azzi S, Abi Habib W, Netchine I. Beckwith-Wiedemann
and Russell-Silver Syndromes: from new molecular insights
to the comprehension of imprinting regulation. Curr Opin
Endocrinol Diabetes Obes 2014;21:30-8. Crossref
11. Price SM, Stanhope R, Garrett C, Preece MA, Trembath
RC. The spectrum of Silver-Russell syndrome: a clinical
and molecular genetic study and new diagnostic criteria. J
Med Genet 1999;36:837-42.
12. Bartholdi D, Krajewska-Walasek M, Ounap K, et al.
Epigenetic mutations of the imprinted IGF2-H19 domain
in Silver-Russell syndrome (SRS): results from a large
cohort of patients with SRS and SRS-like phenotypes. J
Med Genet 2009;46:192-7. Crossref
13. Eggermann T, Gonzalez D, Spengler S, Arslan-Kirchner M,
Binder G, Schönherr N. Broad clinical spectrum in Silver-Russell syndrome and consequences for genetic testing in
growth retardation. Pediatrics 2009;123:e929-31. Crossref
14. Dias RP, Nightingale P, Hardy C, et al. Comparison of
the clinical scoring systems in Silver-Russell syndrome
and development of modified diagnostic criteria to guide
molecular genetic testing. J Med Genet 2013;50:635-9. Crossref
15. Hitchins MP, Stanier P, Preece MA, Moore GE. Silver-Russell syndrome: a dissection of the genetic aetiology and
candidate chromosomal regions. J Med Genet 2001;38:810-9. Crossref
16. Fuke T, Mizuno S, Nagai T, et al. Molecular and clinical
studies in 138 Japanese patients with Silver-Russell
syndrome. PLoS One 2013;8:e60105. Crossref
17. Bliek J, Terhal P, van den Bogaard MJ, et al. Hypomethylation
of the H19 gene causes not only Silver-Russell syndrome
(SRS) but also isolated asymmetry or an SRS-like
phenotype. Am J Hum Genet 2006;78:604-14. Crossref
18. Bruce S, Hannula-Jouppi K, Peltonen J, Kere J, Lipsanen-Nyman M. Clinically distinct epigenetic subgroups in Silver-Russell syndrome: the degree of H19 hypomethylation
associates with phenotype severity and genital and skeletal
anomalies. J Clin Endocrinol Metab 2009;94:579-87. Crossref
19. Kotzot D. Maternal uniparental disomy 7 and Silver-Russell syndrome—clinical update and comparison with
other subgroups. Eur J Med Genet 2008;51:444-51. Crossref
20. Binder G, Seidel AK, Martin DD, et al. The endocrine
phenotype in Silver-Russell syndrome is defined by the
underlying epigenetic alteration. J Clin Endocrinol Metab
2008;93:1402-7. Crossref
21. Azzi S, Salem J, Thibaud N, et al. A prospective study
validating a clinical scoring system and demonstrating
phenotypical-genotypical correlations in Silver-Russell
syndrome. J Med Genet 2015;52:446-53. Crossref
22. But WM, Huen KF, Lee CY, Lam YY, Tse WY, Yu CM. An
update on the indications of growth hormone treatment
under Hospital Authority in Hong Kong. Hong Kong J
Paediatr 2012;17:208-16.
23. Azzi S, Blaise A, Steunou V, et al. Complex tissue-specific
epigenotypes in Russell-Silver Syndrome associated with
11p15 ICR1 hypomethylation. Hum Mutat 2014;35:1211-20. Crossref
24. Bullman H, Lever M, Robinson DO, Mackay DJ, Holder
SE, Wakeling EL. Mosaic maternal uniparental disomy of
chromosome 11 in a patient with Silver-Russell syndrome.
J Med Genet 2008;45:396-9. Crossref
25. Brioude F, Oliver-Petit I, Blaise A, et al. CDKN1C mutation
affecting the PCNA-binding domain as a cause of familial
Russell Silver syndrome. J Med Genet 2013;50:823-30. Crossref
26. Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH,
Temple IK. Temple syndrome: improving the recognition
of an underdiagnosed chromosome 14 imprinting
disorder: an analysis of 51 published cases. J Med Genet
2014;51:495-501. Crossref
27. Kagami M, Mizuno S, Matsubara K, et al. Epimutations
of the IG-DMR and the MEG3-DMR at the 14q32.2
imprinted region in two patients with Silver-Russell
Syndrome–compatible phenotype. Eur J Hum Genet
2015;23:1062-7. Crossref
28. Hämäläinen RH, Mowat D, Gabbett MT, O’brien TA,
Kallijärvi J, Lehesjoki AE. Wilms’ tumor and novel TRIM37
mutations in an Australian patient with mulibrey nanism.
Clin Genet 2006;70:473-9. Crossref
29. van der Wal G, Otten BJ, Brunner HG, van der Burgt I. 3-M
syndrome: description of six new patients with review of
the literature. Clin Dysmorphol 2001;10:241-52. Crossref
30. Scott RH, Douglas J, Baskcomb L, et al. Methylation-specific
multiplex ligation-dependent probe amplification
(MS-MLPA) robustly detects and distinguishes 11p15
abnormalities associated with overgrowth and growth
retardation. J Med Genet 2008;45:106-13. Crossref
31. Spengler S, Begemann M, Ortiz Brüchle N, et al. Molecular
karyotyping as a relevant diagnostic tool in children with
growth retardation with Silver-Russell features. J Pediatr
2012;161:933-42. Crossref